胃蛋白酶亲和有机聚合物毛细管整体柱的制备及性能考察
2014-08-03池翠杰季一兵
池翠杰, 王 伟, 季一兵
(中国药科大学理学院,江苏南京210009)
毛细管电色谱(CEC)具有毛细管电泳(CE)和高效液相色谱(HPLC)的双重分离性能[1],被广泛用于手性药物、小分子物质、生物样品以及复杂混合物的分离分析[2-7]。毛细管整体柱具有制备方法简单、化学性质稳定、传质效率高、柱容量高等优点,被誉为第4代分离介质[8]。聚丙烯酸酯有机聚合物整体柱,因聚合单体选材范围广,可灵活在固定相中引入需要的活性基团,且使用的pH范围宽,在pH 2~12范围内能稳定存在,而成为目前研究最普遍的CEC 固定相基质之一[9]。
蛋白质类手性选择剂因独特的三维结构而对手性对映体有强大的拆分能力[10]。限于蛋白质作添加剂时背景吸收会干扰分析测定[11],因此其作为固定相的应用得到迅速发展[12-14]。但是应用于整体柱CEC手性固定相中的报道目前仅限于牛血清白蛋白(BSA)和卵粘蛋白(OVM)[15,16],且拆分的药物非常有限,只实现了色氨酸等氨基酸、华法林、安息香的分离,蛋白质亲和整体柱CEC手性拆分的优势没有得到充分发挥。因此探寻拆分能力更强的蛋白质手性选择剂,以CEC整体柱为基质制备手性固定相(CSP)是目前较有前景的研究方向。
胃蛋白酶是一种酸性蛋白质,相对分子质量为34 600,等电点约为1,由327个氨基酸组成。其作为手性选择剂适合拆分碱性和中性的手性对映体,具有广泛的应用范围,已成功应用于HPLC和CE对各类光学异构体的分离[17-19]。Fanali等[17]将胃蛋白酶作为手性添加剂应用于毛细管区带电泳(CZE)实现了普萘洛尔、异丙嗪、维拉帕米等的拆分。Haginaka等[18]将胃蛋白酶键合在硅胶颗粒上,制备填充HPLC柱,实现了苯二氮卓艹类、抗组胺类、β-阻断剂类等25种药物的拆分,充分体现了胃蛋白酶强大的手性拆分能力。随后Haginaka等[19]又制备了胃蛋白酶和卵类粘蛋白混合手性HPLC固定相,对异丙基肾上腺素和阿普洛尔具有拆分效果。而以整体柱为基质制备胃蛋白酶的CSP在CEC中的应用还未见报道。将胃蛋白酶强大的拆分性能和整体柱的优势相结合会为亲和CEC的手性拆分提供新的思路。
本文合成了聚(甲基丙烯酸缩水甘油酯(glycidyl methacrylate,GMA)-乙二醇二甲基丙烯酸酯(ethyleneglycol dimethacrylate,EDMA))毛细管整体柱,探讨了聚合反应条件的影响,优化了基质柱的制备条件。对合成的整体柱进行了性能考察,在加压毛细管电色谱模式下将其用于苯等小分子化合物的分离。以聚(GMA-EDMA)整体柱为基质,戊二醛为连接臂制备了胃蛋白酶亲和整体柱,并应用于碱性药物的拆分,为有机聚合物整体柱对小分子物质以及蛋白质亲和CEC对手性药物的分离分析提供了方法。
1 实验部分
1.1 仪器与试剂
HP3DCE 7100毛细管电泳仪(美国Agilent公司),S-3400NⅡ扫描电子显微镜(日本Hitachi公司);Agilent 1200高效液相色谱仪(美国Agilent公司);ODGC-14B气相色谱柱温箱(日本Shimadzu公司);pHs-25型pH计(上海精密科学仪器有限公司);电子分析天平(美国 Sartorius公司);SWQ-IA智能数字恒温水浴锅(南京桑力电子设备厂)。未涂层石英毛细管柱(75 μm I.D.,365 μm O.D.,河北永年光导纤维厂)。GMA、EDMA、γ-甲基丙烯酸氧丙基三甲氧基硅烷(γ-MAPS)(纯度98%,美国 Sigma公司);正丙醇、1,4-丁二醇、戊二醛、氰基硼氢化钠(Aladdin公司);偶氮二异丁腈(AIBN)(上海第四试剂厂);胃蛋白酶(美国Sigma公司);奈福泮、氨氯地平、西酞普兰、扑尔敏均为消旋体;醋酸(HAc)、醋酸铵(NH4Ac)和其余试剂均为分析纯;实验用水为纯净水。上述液体试剂在使用前用0.22 μm有机微孔滤膜过滤,GMA和EDMA使用前用0.1 mol/L NaOH溶液处理以除去阻聚剂。
1.2 方法
1.2.1 毛细管内壁的预处理
毛细管内壁依次用1 mol/L的NaOH溶液和0.1 mol/L的HCl溶液冲洗,然后用水冲洗干净,N2吹干。将含50%(体积分数)γ-MAPS的甲醇溶液注入毛细管中,毛细管两端封口,于50℃水浴中反应12 h,然后将毛细管置于氮吹装置上,在0.2 MPa下用甲醇冲洗2 h,N2吹干,备用。
1.2.2 聚(GMA-EDMA)毛细管整体柱的制备
毛细管整体柱反应液由活性单体GMA,交联剂EDMA,致孔剂体系1,4-丁二醇、正丙醇和引发剂AIBN组成。GMA和EDMA的质量比为3∶2,GMA和EDMA质量之和占35%,正丙醇和1,4-丁二醇的质量比为3∶1,AIBN的质量分数为1.0%。将反应液混匀,通氮气除氧,过滤,灌注于经前处理的毛细管(毛细管总长为35.0 cm,有效长度为25.0 cm)中,两端封口,于44℃水浴中反应12 h。依次用甲醇、水冲洗至整体柱内的致孔剂、未反应的单体和其他杂质去除完全,保存备用。
1.2.3 胃蛋白酶亲和手性整体柱的制备
于氮吹装置0.4 MPa恒压条件下制备胃蛋白酶亲和手性整体柱。取4 mL质量分数为25%的浓氨水,加1 mL水,配制成质量分数为20%的氨水溶液,灌入整体柱,于40℃反应5 h。取1 mL质量分数为50%的戊二醛溶液,加入4 mL 100 mmol/L磷酸盐溶液(pH 8.0),配制成质量分数为10%的戊二醛磷酸盐溶液,不断通入柱中,避光反应5 h。室温下以5 mg/mL胃蛋白酶反应溶液(含氰基硼氢化钠2.5 mg/mL)冲柱12 h,用100 mmol/L NH4Ac-HAc溶液(pH 4.5)冲柱2 h。用电色谱条件下的流动相(15 mmol/L NH4Ac-HAc(pH 5.0))平衡1 h。
1.2.4 整体柱压力测定
将毛细管一端连接在高效液相色谱仪上,以甲醇或水为流动相,设定一定流速,使流动相从毛细管的另一端流出,待压力显示稳定时记录为柱压。
1.2.5 电色谱条件
在靠近毛细管整体柱末端处烧制2 mm左右的检测窗口,检测窗口与整体柱出口端的距离为1.5 cm。整体柱在分离样品前,用电色谱条件下待运行的流动相平衡至基线平稳。苯同系物的检测波长为210 nm,氨氯地平、西酞普兰的检测波长分别为240、235 nm,奈福泮、扑尔敏的检测波长为215 nm。
1.2.6 标准溶液的配制
分别取奈福泮、氨氯地平、西酞普兰、扑尔敏4种药物的消旋体,精密称定,甲醇溶解,分别配制成1 mg/mL的溶液。
2 结果与讨论
2.1 聚(GMA-EDMA)整体柱制备条件的优化
尝试使用环己醇-十二烷醇致孔剂系统,结果表明制备的整体柱收缩现象较严重,稳定性和重现性较差,不利于条件的控制,且溶液黏度较大,导致反应液不易混匀。故选用溶液黏度适中的脂肪族醇类正丙醇-1,4 丁二醇致孔剂系统[20]。
考察了单体、交联剂、致孔剂及温度对整体柱的柱压和渗透性的影响,如表1所示。结果表明:柱压随正丙醇和1,4-丁二醇质量比的增加而减小。这是因为正丙醇在反应系统中属于不良溶剂,使得相分离发生较早,形成的聚合物小球较大,其间的空隙也较大,渗透性良好。柱压随GMA和EDMA质量比的减小而减小。EDMA含量增加,反应初期会出现高度交联的聚合物,而使相分离发生较早,这和溶解性差的致孔剂正丙醇效果相似,但交联剂含量增加会使得交联度增加,机械强度较大。柱压随GMA和EDMA总量的增加而增加。根据相分离原理,致孔剂含量越高,孔隙率越大,渗透性越好。温度越高,柱压越大。温度主要影响AIBN活性,温度高则产生自由基数量多,孔隙小[21]。
综合考虑渗透性、机械性能和环氧活性基团含量,初步选定反应物配比如1.2.2节所述。固定上述条件,在40~50℃间调节温度,优选适合实验室氮吹装置操作的整体柱。通过实验,最终确定整体柱的反应温度为44℃。
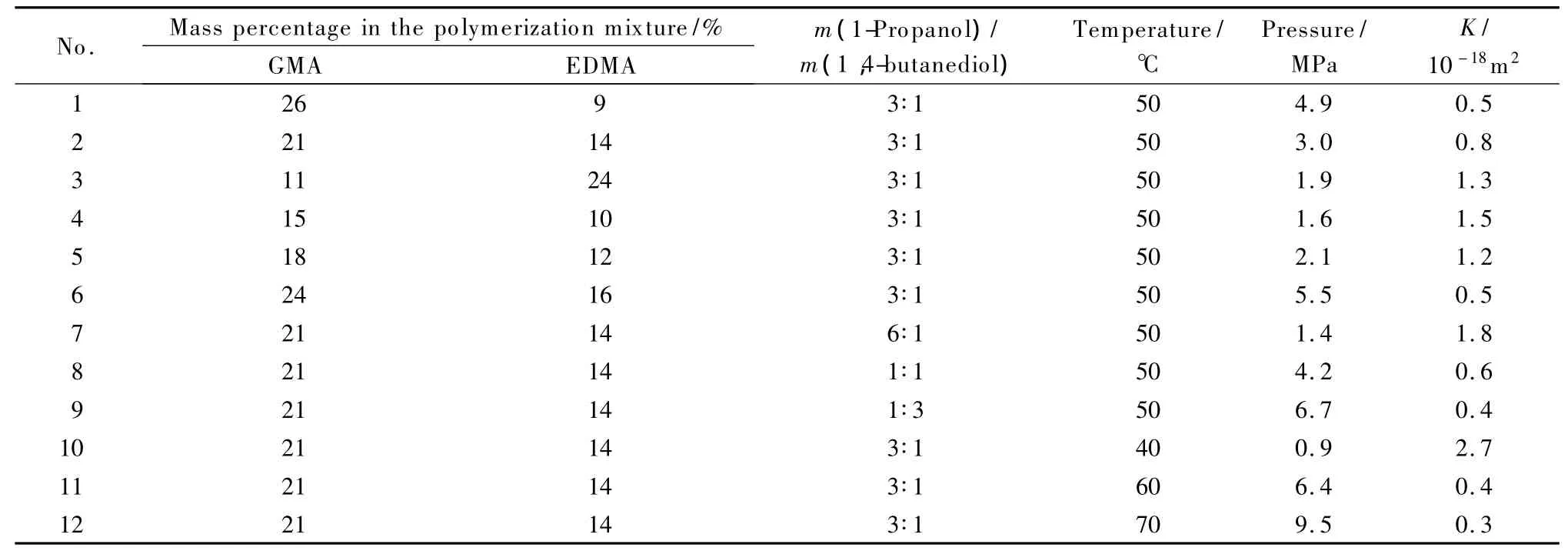
表1 单体、交联剂、致孔剂及温度对整体柱的影响Table 1 Effects of the monomer,cross-linking agent,solvent and temperature on the monolithic column
2.2 聚(GMA-EDMA)整体柱的性能考察
2.2.1 整体柱内的微观结构
用扫描电子显微镜(SEM)观察柱内部形貌(见图1a~c),可见其放大50万倍、5万倍和1万倍的断面结构微孔。在优化的实验条件下制备的整体柱结构均匀,柱内同时存在3种孔结构:一种是小颗粒与小颗粒之间形成的微孔;另一种是小颗粒与“簇”之间形成的中孔;还有一种是“簇”与“簇”之间形成的大孔,既保证了整体柱有良好的渗透性,又可以使溶质和固定相之间充分地相互作用。
图1d为胃蛋白酶亲和毛细管整体柱的扫描电镜图,与键合前的聚(GMA-EDMA)整体柱并无显著差别,说明经过一系列的反应后整体柱的基质结构和孔径依然均匀分布,没有发生塌陷、溶胀等。

图1 整体柱的扫描电镜图Fig.1 Scanning-electron micrographs of the monolithic column
2.2.2 整体柱的耐压性和渗透性考察
以甲醇为流动相,考察聚(GMA-EDMA)整体柱的耐压性和渗透性。柱背压与流动相流速的关系如图2所示。结果显示柱背压与流动相流速的线性关系良好(r2>0.99)。实验操作中的压力在整体柱稳定范围内,柱机械强度和抗压性良好。整体柱的渗透率为2.05×10-18m2,说明其渗透性良好。
以水作为流动相考察胃蛋白酶亲和整体柱的柱压和流速的关系,并比较胃蛋白酶键合反应前后整体柱渗透性的变化。由图2看出键合胃蛋白酶后整体柱压力和线速度仍保持良好的线性关系(r2>0.99)。渗透率为1.93×10-18m2,略小于聚(GMAEDMA)基质柱。
2.2.3 烷基苯类化合物的分离
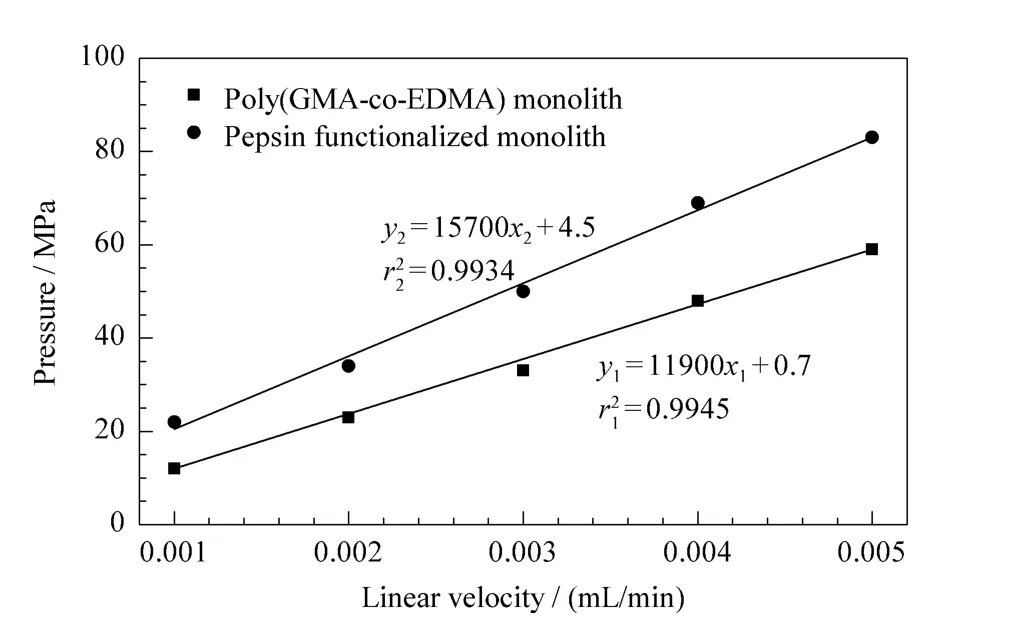
图2 整体柱柱压与流速的线性关系Fig.2 Linear dependency of the back pressure of monolith on the linear velocity of eluent
中性化合物在电色谱中的保留主要是通过溶质分子和固定相间的色谱作用。聚(GMA-EDMA)整体柱基质在电色谱条件下电渗流非常小,使分析物出峰时间过长,故使用所制备的毛细管整体柱在加压电色谱条件下分离烷基苯类化合物。因加压CEC模式和高效液相色谱类似,压力是推动流体前进的驱动力之一。由烷基苯同系物在此整体柱上的电色谱分离情况(见图3)可知,烷基苯同系物按照疏水性由小到大的顺序依次洗脱;分离度均大于2,峰形良好。9.0%,重复性良好。不同批次间整体柱上分析物保留因子的RSD均不大于7.9%,分离度的RSD均不大于5.6%。
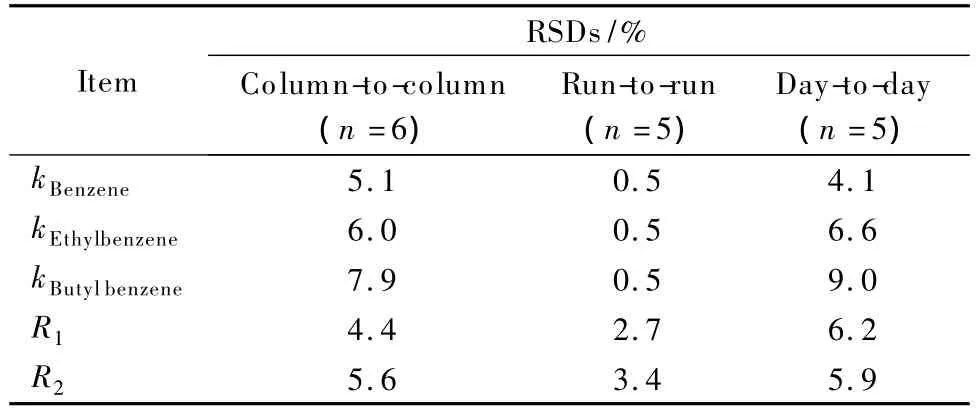
表2 保留因子和分离度的稳定性和重复性考察Table 2 Reproducibilities and stabilities of retention factors and resolutions
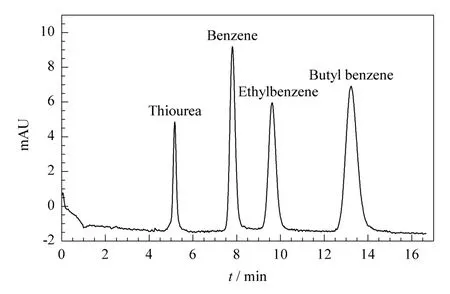
图3 烷基苯同系物在聚(GMA-EDMA)整体柱上的分离Fig.3 Separation of alkyl benzenes compounds on poly(GMA-co-EDMA)monolith
图4为乙腈含量对烷基苯同系物保留行为的影响。苯同系物的保留因子(k)随着流动相中乙腈含量的增加而降低,lg k与流动相中乙腈含量具有良好的线性关系(r2>0.98),说明中性化合物在该整体柱上的保留类似于在反相高效液相色谱上,整体柱表现出了典型的反相色谱特征。
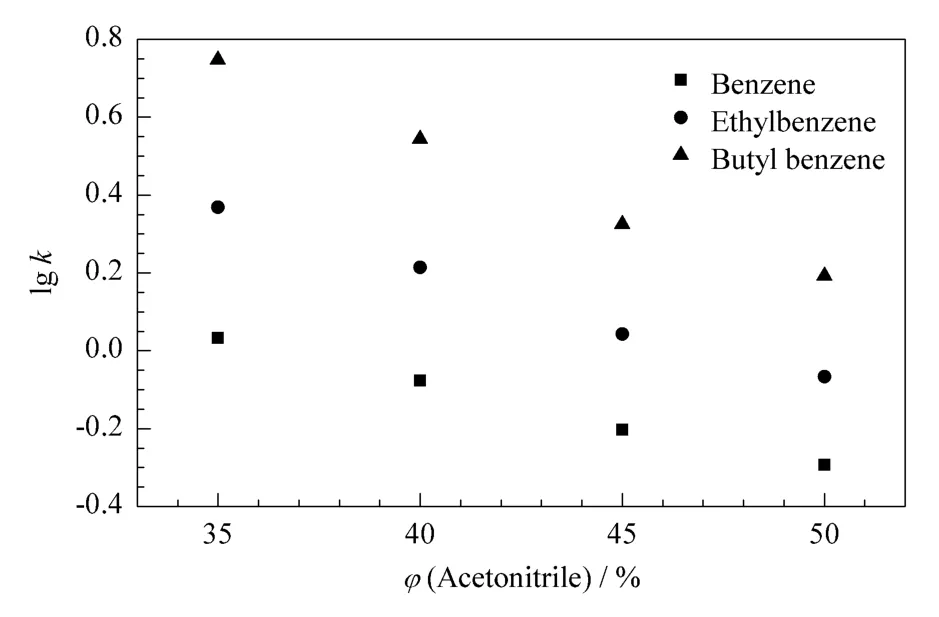
图4 乙腈含量对烷基苯同系物保留行为的影响Fig.4 Effect of acetonitrile content on the retention of alkyl benzenes
2.2.4 整体柱稳定性和重现性考察
以硫脲为标记,分离苯、乙苯、丁基苯混合物,考察毛细管整体柱的重复性,结果见表2。不同分析物保留因子的日内精密度(RSD)均为0.5%,分离度的RSD不大于3.4%;日间精密度均不大于
2.3 胃蛋白酶手性整体柱固定相的性能考察
2.3.1 pH值对电渗流和手性药物分离的影响
本实验中整体柱的电渗流(EOF)主要是由胃蛋白酶解离产生,电渗流大小与所用溶液的pH相关。实验考察了流动相中缓冲液的pH对EOF的影响。从图5可以看出,在pH 4.0~6.0范围内整体柱电渗流方向均为正向,且随着pH的增大而增大。
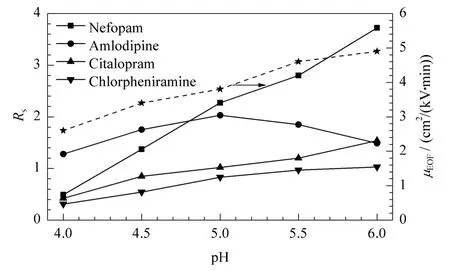
图5 pH对电渗流(EOF)和对映体分离度(Rs)的影响Fig.5 Effect of pH on electroosmotic flow(EOF)and resolution(Rs)of the enantiomers
pH值会影响分析物的质子化程度及固定相蛋白质的离解程度,进而影响分析物与手性固定相的相互作用力及电渗流的大小,从而影响分离。pH大于6.0时胃蛋白酶不稳定,因此在pH 4.0~6.0范围内,考察了其对奈福泮、氨氯地平、西酞普兰、扑尔敏手性拆分的影响。结果表明:氨氯地平的分离度随着pH的增大而增大,pH 5.0时达到最大,随后随pH的增大而减小;奈福泮、西酞普兰和扑尔敏的分离度均随着pH的增大而增大,在pH 6.0时达到最大。
2.3.2 运行电压对手性药物分离的影响
考察运行电压从10 kV到25 kV变化对奈福泮和氨氯地平对映体分离的影响。结果显示,奈福泮对映体的分离度随着电压升高而降低,电压由10 kV增加到25 kV,分离度由3.72降低到1.49。而氨氯地平对映体的分离度随着电压增加先增加后减小,在电压为20 kV时达到分离度最大,为2.03。
在分离的pH(分别为5.0和6.0)条件下,整体柱具有正向的电渗流,说明整体柱带负电荷。电渗流产生是因pH大于胃蛋白酶的等电点,其带净电荷为负。整体柱内存在电荷主要为羧基电离产生的负电荷,同时存在氨基电离产生的正电荷。两种对映体的pKa接近10.0,在分离的pH条件下带正电荷,因此与整体柱间存在静电作用;另外,整体柱基质本身具有反相色谱特征,与含有苯环的药物间存在疏水作用力。
对映体运行的驱动力除了电渗流外还有自身的电泳迁移。药物的保留和分离是在上述各因素的综合作用下实现的。电压增加,药物的电泳速度增大,对映体在整体柱内保留时间缩短,与整体柱间的各种作用力的作用时间和强度均减小。
因药物的结构及性质不同,导致其疏水性、离解性均不同,导致分离度随实验条件变化的趋势不同。
2.3.3 温度和进样量对手性药物分离的影响
以奈福泮为例,考察温度对分离的影响。由表3可知随着温度升高,奈福泮的分离度和选择因子均降低,保留时间减小,柱效增加。因CEC同时具有高效液相色谱和电泳的双重保留机理,温度升高时溶液黏度减小,电渗流增加,驱动力增加,因此奈福泮保留时间减小,而对映体与蛋白质间的手性“3点作用力”的作用时间因为保留时间缩短而缩短,因对映体均存在色谱保留,即非水作用力时间也减弱,故而分离度减小而柱效增加。
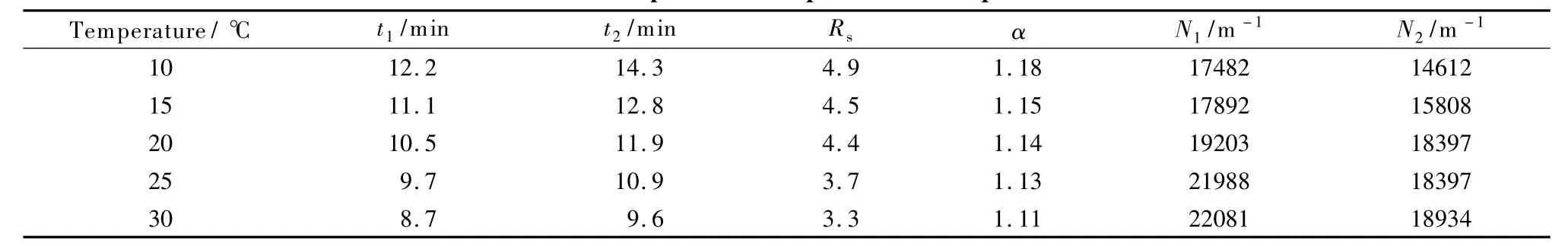
表3 温度对奈福泮对映体分离的影响Table 3 Effect of temperature on separation of nefopam enantiomers
以奈福泮分离为例考察进样量对手性药物分离情况的影响。电压方式进样(5 kV×1 s降至1 kV×1 s),分离结果表明,减小进样量时奈福泮的分离度由1.29提高到4.60。进样量对分离效果影响很大。可能的原因是药物分子间相互竞争手性选择剂蛋白质的作用位点,而进样量小时,对映体与固定数量的有效手性作用位点作用相对更充分,而使分离度增加。
2.3.4 胃蛋白酶亲和手性整体柱对药物的分离
胃蛋白酶亲和手性整体柱能够实现对奈福泮、氨氯地平、西酞普兰、扑尔敏的拆分,前3种药物都能实现基线分离(见图6)。
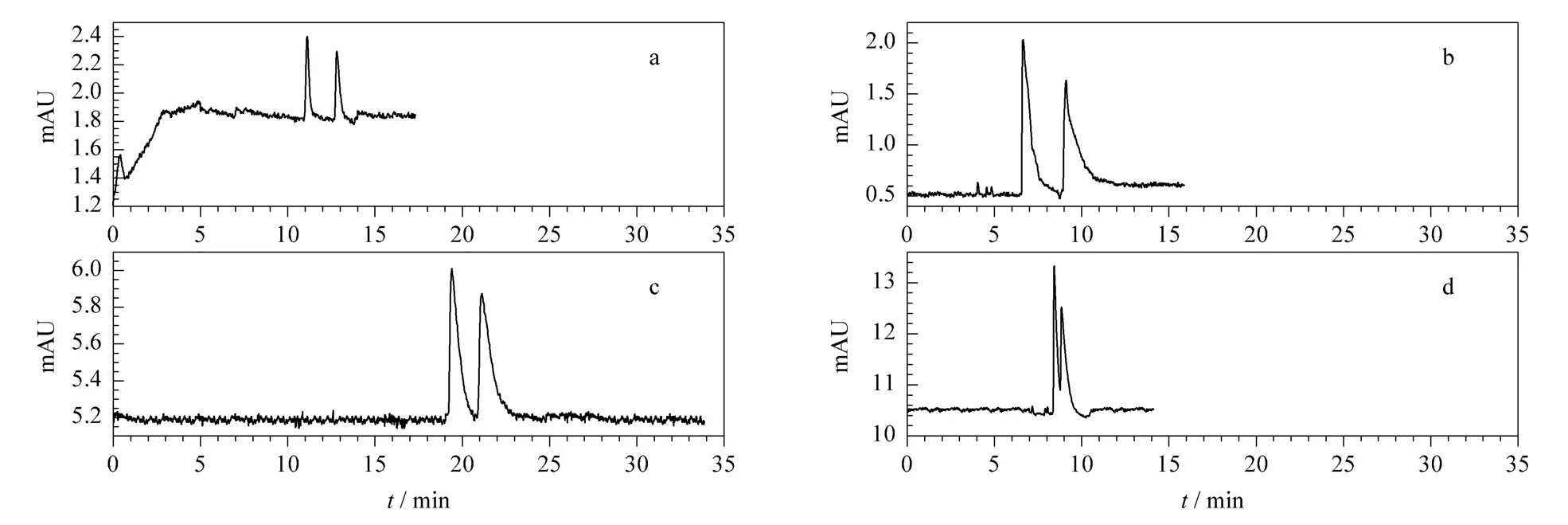
图6 胃蛋白酶亲和手性整体柱分离4种手性药物的色谱图Fig.6 Enantioseparation chromatograms of(a)nefopam,(b)amlodipine,(c)citalopram and(d)chlorpheniramine on pepsin-bonded chiral monolithic column
3 结论
探讨了聚(GMA-EDMA)毛细管整体柱合成的影响因素,优化了整体柱制备条件。所制备的整体柱内部结构均匀、柱渗透性、机械强度良好。在加压电色谱模式下将整体柱用于小分子物质的分离分析,柱效和分离度较高,重现性和稳定性良好,其保留行为具有典型的反相色谱特征,对小分子化合物的分离分析具有指导意义。以戊二醛为连接臂制备了胃蛋白酶亲和手性整体柱,在电色谱条件下应用于手性药物的拆分。本研究为蛋白质亲和CEC的制备和应用提供了新的思路和方法。
[1] Yue C Y,Ding G S,Tang A N.Chinese Journal of Chromatography(岳春月,丁国生,唐安娜.色谱),2013,31(1):10
[2] Lin J,Liu S F,Lin J,et al.J Chromatogr A,2011,1218(29):4671
[3] Gao Y,Wang Y,Wang C R,et al.Chinese Journal of Chromatography(高也,王彦,王超然,等.色谱),2012,30(5):487
[4] Tian Y,Zhong C,Fu E Q,et al.J Chromatogr A,2009,1216(6):1000
[5] Li Y J,Song C H,Zhang L Y,et al.Talanta,2010,80(3):1378
[6] Liu W,Qi J R,Yan L N,et al.J Chromatogr B,2011,879(28):3012
[7] Bai L G,Niu W J,Yang G L.Chinese Journal of Chromatography(白立改,牛文敬,杨更亮.色谱),2013,31(4):303
[8] Han H F,Wang Q,Liu X,et al.J Chromatogr A,2012,1246(13):9
[9] Aguiar V S,Bottoli C B G.Microchem J,2013(109):51
[10] He J,Ji Y B.Pharmaceutical and Clinical Research(何健,季一兵.药学与临床研究),2013,21(2):166
[11] Haginaka J.J Chromatogr A,2000,875(1/2):235
[12] Mallik R,Jiang T,Hage D S.Anal Chem,2004,76(23):7013
[13] Hage D S,Anguizola J A,Bi C,et al.J Pharmaceut Biomed,2012,69:93
[14] Yao C H,Qi L,Qiao J,et al.Talanta,2010,82(4):1332
[15] Kato M,Sakai-Kato K,Matsumoto N,et al.Anal Chem,2002,74(8):1915
[16] Zheng Y,Lei W,Zhang L Y,et al.Journal of East China University of Science and Technology(郑翌,雷雯,张凌怡等.华东理工大学学报),2011,37(2):199
[17] Fanali S,Caponecchi G,Aturki Z.J Microcolumn Sep,1997,9(1):9
[18] Haginaka J,Miyano M,Saizen Y,et al.J Chromatogr A,1995,708:161
[19] Haginaka J,Miyano Y.Anal Sci,1996,12:727
[20] Grafnetter J,Coufal P,Tesarova E,et al.J Chromatogr A,2004,1049(1/2):43
[21] Zou J J.[MS Dissertation].Nanjing:Nanjing University of Science and Technology(邹娟娟.[硕士学位论文].南京:南京理工大学),2004:24
