铈离子与H2O和CO2分子相互作用的理论研究
2014-07-31罗冬梅
罗冬梅
(赤峰学院 化学系, 内蒙古 赤峰024000)
铈离子与H2O和CO2分子相互作用的理论研究
罗冬梅
(赤峰学院 化学系, 内蒙古 赤峰024000)

罗冬梅, 女,1976年出生于黑龙江省,1999年7月毕业于齐齐哈尔大学化学系,2007年7月毕业于南昌大学理学院,获物理化学专业硕士学位。2010年7月毕业于南昌大学理学院,获工业催化专业博士学位。主要从事光催化和分子中药学的基础理论研究。
本文对Ce负载TiO2光催化水还原CO2制取甲醇的理论研究,做了部分工作.采用范德华配合物模型,利用高斯03软件包,在B3LYP水平上研究了Ce离子得电子前后分别与反应物分子相互作用的情况,得到了相应的几何参数和电荷分布以及相互作用的能量数据,计算结果表明Ce对反应物有影响,Ce活化了H2O和CO2分子.其中4价的铈对H2O和CO2分子的影响更大.比较H2O和CO2与Ce离子在基态和激发态时的相互作用,发现相对高价的金属离子对水和二氧化碳分子有较强的活化作用,激发态的配合物相互作用能量更高,但几何参数与基态比较变化不大.说明铈离子在反应体系中总体上是高价态时基态对反应物分子的活化能力强,低价时激发态相互作用能较大,说明铈离子可能参与了光生电子的传输.
铈离子;CO2;H2O;相互作用
1 前言
自从人类进入工业化生产以来,煤炭、石油的大量使用,空气中的二氧化碳含量一直在上升,温室效应严重影响着生态环境,因此解决其排放问题很重要.CO2减排对于控制全球变暖趋势问题上起到关键作用,如何利用空气中十分丰富的二氧化碳作为碳资源是许多科学家一直想解决的问题.自从Inoue et al.首次报道[1]光催化H2O还原CO2以来,光催化水还原CO2的研究受到广泛关注.由二氧化碳制备甲醇,不仅可利用自然界中廉价而丰富的碳资源合成重要的化工产品,同时减轻二氧化碳对环境所造成的负面影响.其中,CO2的固定再利用技术为新能源的开发提供了一条新的途径[2-3].
甲醇是重要的有机化工原料,近年来,人们对CO2和H2O制备甲醇反应的研究也越来越多,已使之成为较热门的研究课题之一.计算机分子模拟量子化学和密度泛函理论的发展,使我们对实验可行性以及新型材料性能的预测成为可能.近年来,计算机分子模拟量子化学和密度泛函理论的发展使之成为可能.利用计算机模拟真实材料系统与外界条件的相互作用.
为深入了解反应的微观过程和机理,研究反应物和产物与催化剂及其表面负载金属离子的相互作用,了解反应物的活化.采用简化的范德华模型体系,理论上定性研究预测光催化反应过程中活性组分与反应物分子的相互作用,既可以节省筛选催化剂的时间又可以解释活性物种的作用,并为反应机理的探讨提供一定依据.本文选取负载于二氧化钛的铈离子,研究其与产物分子的相互作用,采用的配合物模型为Ce(+4)–2H2O–CO2和Ce(+3)–2H2O–CO2.
2 计算细节
采用G03[4]程序包,在B3LYP[5]水平上,由于大的基组对计算电子亲和力有用而对键长和键角不是很有用[6],因而O, C和H选取6-31G[7]基组,稀土金属离子Ce采用Stuttgart RSC 1997 ECP EMSL基组[8]进行几何优化.DFT方法能够直接确定精确的基态能量和电子密度,可以大为简化电子结构的计算.本文采用的是常用的杂化交换和相关能量泛函:
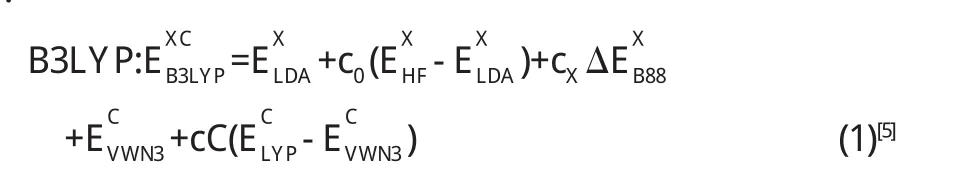
3 Ce(+4)和Ce(+3)与H2O和CO2的相互作用
铈离子与反应物分子的相互作用采用范德华配合物模型:Ce(+4)-2H2O–CO2和Ce(+3)–2H2O–CO2,在B3LYP水平上计算完成,得到相应的优化的结构参数,和相互作用能量.图3-1(a)为Ce(+4)–2H2O–CO2和Ce(+3)–2H2O–CO2的优化几何结构,(b)为Ce(+4)–3CO2和Ce(+3)–3CO2的优化几何结构,(c)为Ce(+4)–2H2O和Ce(+3)–2H2O的优化几何机构.
H2O和CO2与Ce(+4)和Ce(+3)分别相互作用.选取两种配合物模型M–3CO2和M–2H2O–CO2(M=Ce(+4)和Ce (+3))研究Ce(+4)和Ce(+3)与H2O和CO2的相互作用.优化的几何参数如图3-1和下表3-1、3-2、3-3所示,结果表明CO2和H2O的键长都有所变化.Ce(+4)–2H2O–CO2的C=O键是1.151Å和1.262Å,在Ce(+3)–2H2O–CO2中,C=O键是1.151Å和1.235Å,而CO2的为1.188Å.Ce(+4)–2H2O–CO2中的H–O键是1.009Å,AH-O-H是105.1°在Ce(+3)–2H2O–CO2中H–O键是0.9861Å,AH-O-H是106.5°,而H2O中的H–O键为0.9699Å,AH-O-H为108.4°.在M–2H2O–CO2模型中,Ce(+4)和Ce(+3)对H2O和CO2都有一定的影响和作用,使得CO2的键长和H2O的键长键角发生了变化,其中Ce(+4)对CO2和H2O的作用更大些,活化作用更强.
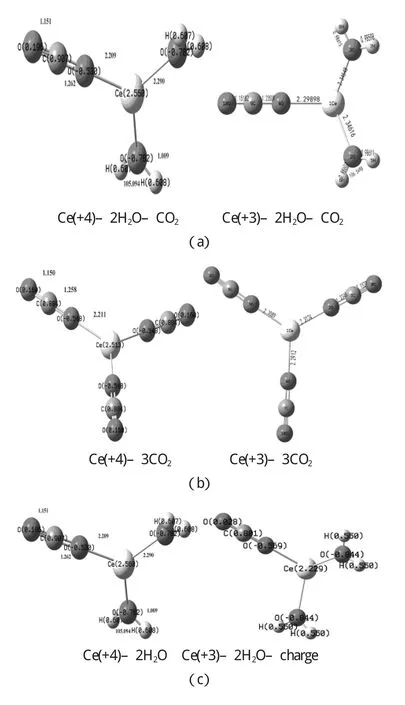

表3-1(Ce(+3)–2H2O–CO2)优化几何结构
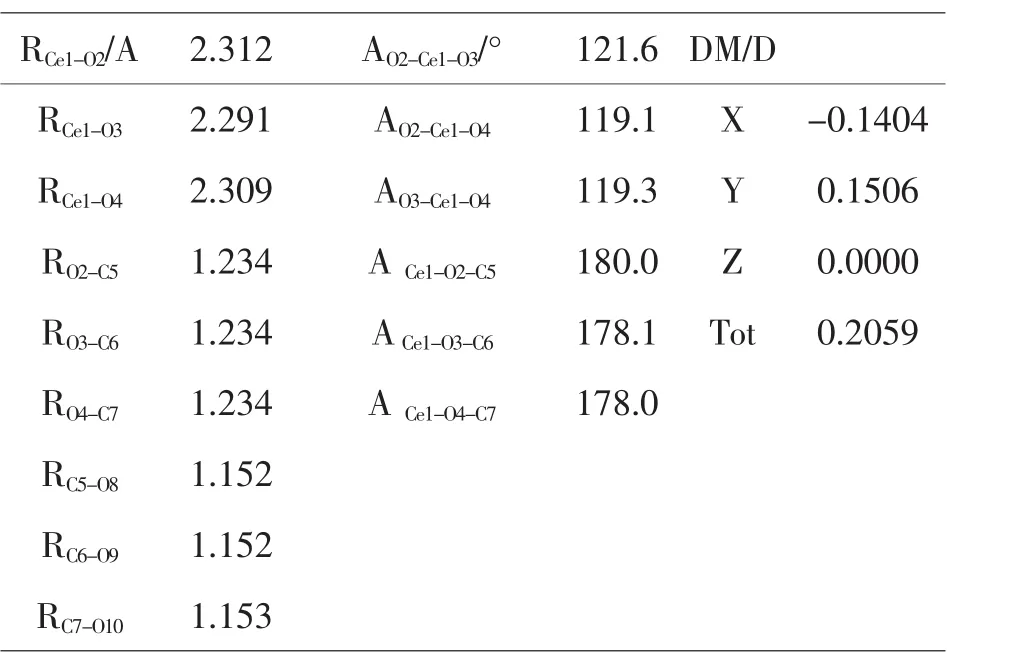
表3 -2(Ce(+3)–3CO2)优化的结构参数和偶极矩
在M–3CO2模型中,Ce(+3)–3CO2的C=O键是1.152Å和1.234Å.Ce(+4)–3CO2的C=O键是1.150Å 和1.258Å,而CO2的为1.188Å.由此可见Ce(+4)对CO2的影响更大,活化作用更强.
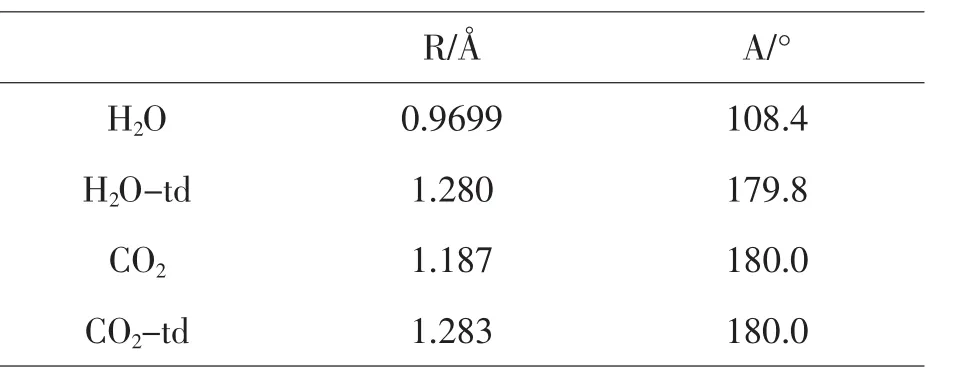
表3 -3水和二氧化碳分子基态和激发态的几何参数
配合物的电荷分布分析如下:
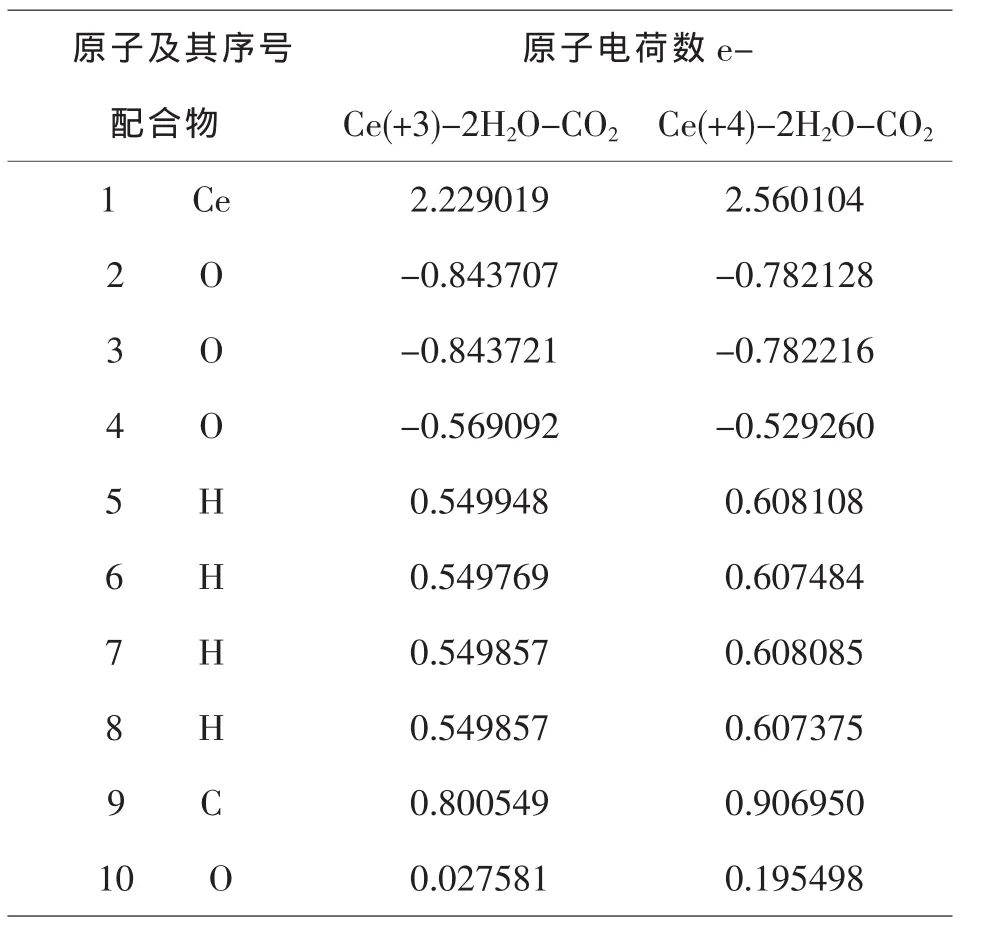
表3 -4 Ce(+3)–2H2O–CO2,Ce(+4)–2H2O–CO2的穆利肯电荷分布
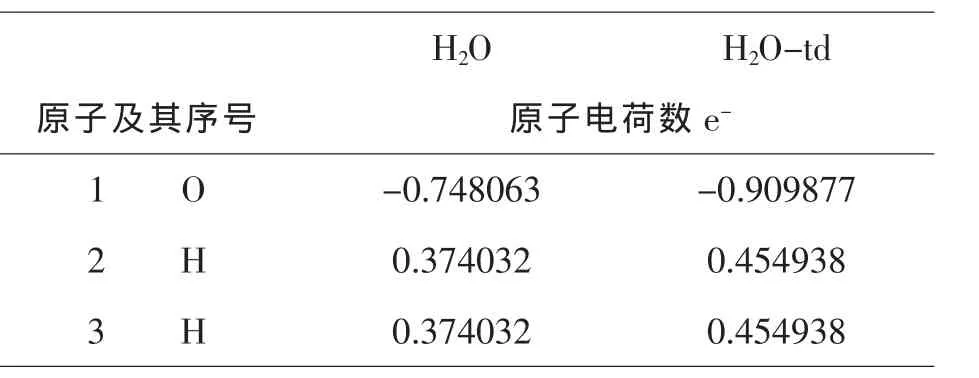
表3 -5水分子基态和激发态的电荷分布
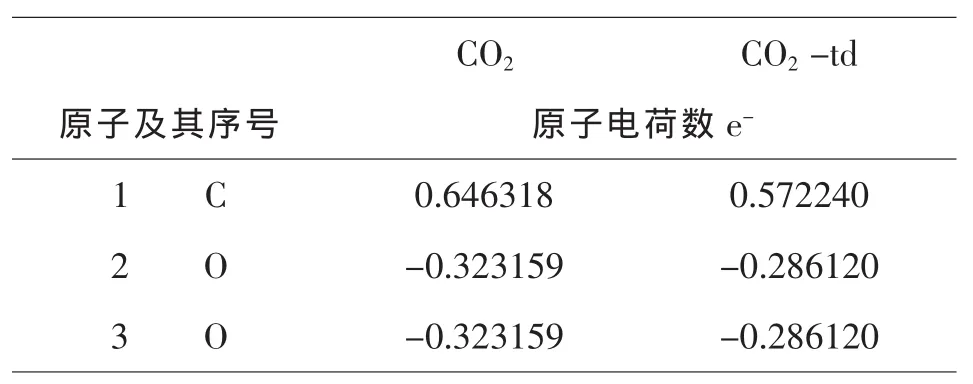
表3 -6二氧化碳分子基态和激发态的穆里立电荷分布
由图3-1和表3-4、3-5、3-6可知,H2O和CO2与Ce(+4)和Ce(+3)分别相互作用后,每个原子的电荷数皆发生变化.由于不同价态铈离子的作用,使得配合物体系中不同原子的电荷分布不同,Ce(+3)的配合物中O(2)原子的电荷为:-0.8437 e-;Ce(+4)的配合物中O(2)原子的电荷为:-0.7821 e-;它们相差了:-0.0616 e-,而且这些分布又不同于单分子水中的O原子的电荷,原因是铈离子的加入使得电场发生了变化.Ce(+3)的配合物中H(5)原子的电荷为0.5499 e-;Ce (+4)的配合物中H(5)原子的电荷为:0.6081 e-;它们相差了:0.0582 e-.除了显负电荷数的O(2,3,4)原子外,配合物中的其他原子的电荷分布均呈现规律性变化,即随着铈离子的价态的升高而增加,O原子的特殊情况是金属离子的电场作用和他自身电负性综合影响的结果.

表3 -7 Ce(+3)–3CO2,Ce(+4)–3CO2的穆立肯电荷分布
由图3-1和表3-5,3-6,3-7可知,CO2与Ce(+4)和Ce (+3)分别相互作用后,每个原子的电荷数皆发生变化.由于不同价态铈离子的作用,使得配合物体系中不同原子的电荷分布不同,Ce(+3)的配合物中O(2)原子的电荷为:-0.5638 e-;Ce(+4)的配合物中O(2)原子的电荷为:-0.5480;它们相差了:-0.0148,而且这些分布又不同于单分子水中的O原子的电荷,原因是铈离子的加入使得电场发生了变化.Ce(+3)的配合物中C(5)原子的电荷为0.8000 e-;Ce(+4)的配合物中C(5)原子的电荷为:0.8835 e-;它们相差了:0.0835 e-.除了显负电荷数的O(2,3,4)原子外,配合物中的其他原子的电荷分布均呈现规律性变化,即随着铈离子的价态的升高而增加,O原子的特殊情况是金属离子的电场作用和他自身电负性综合影响的结果.
相互作用能按照如下公式计算:
该位置是操纵列车常用制动,使列车正常缓慢停车或调整运行速度所使用的位置。包括初制动位和全制动位,两者之间是制动区。
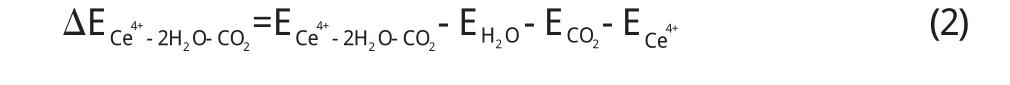

由以上(1)、(2)、(3)、(4)四个式子算出了每个部分的相互作用能,在M–3CO2(M=Ce(+4)和Ce(+3))体系中,Ce (+4)–3CO2的相互作用能为–8.850×108J/mol,而Ce(+3)–3CO2的相互作用能为–8.833×108J/mol.在M–2H2O–CO2(M=Ce(+4)和Ce(+3))体系中,Ce(+4)– 2H2O–CO2的相互作用能为–2.692×106J/mol而Ce(+3)–2H2O–CO2的相互作用为–1.181×106J/mol,由此可见不管是在M–3CO2(M=Ce(+4)和 Ce(+3))体系中还是在M–2H2O–CO2(M=Ce(+4)和Ce(+3))体系中,+4价的Ce与H2O和CO2的相互作用能ΔE较大. ΔE越大,金属离子与反应物分子相互作用力越大,对反应物分子的影响就越大,对反应物分子的活化程度就越大,由此可见Ce(+4)要比Ce(+3)对H2O和CO2影响作用大.
4 基态Ce和激发态Ce与H2O和CO2的相互作用
Ce3+和反应物分子的范德华配合物分别在基态和激发态下在B3LYP水平上完成全优化,得到相应的优化结构参数,电荷分布,和相互作用能量.
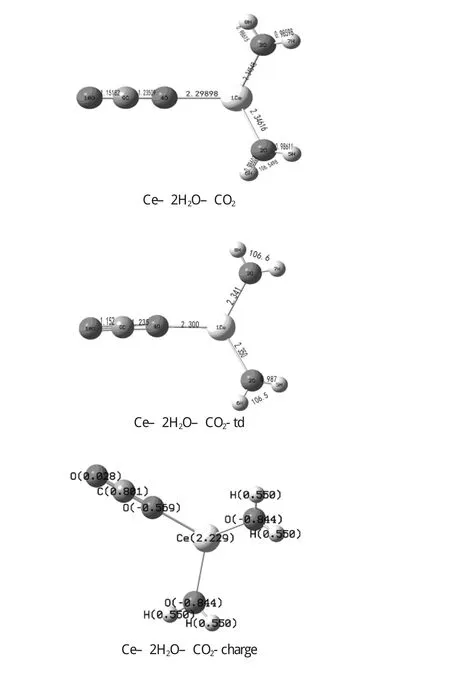

图4 -1激发态优化的Ce-2H2O-CO2几何结构
Ce3+和反应物分子的范德华配合物分别在基态和激发态下在B3LYP水平上完成全优化.选取一种模型M–2H2O–CO2(M=Ce和Ce-td)研究Ce和Ce-td与H2O和CO2的相互作用.优化的几何参数如图4-1和下表9-1.9-2所示,结果表明CO2和H2O的键长皆有所变化.在Ce–2H2O–CO2中的C=O键是1.151Å 和1.235Å,而CO2的为1.188Å.在Ce–2H2O–CO2中的H–O键是0.9861Å 和0.986Å,AH-O-H是106.5°,而H2O中的H–O键为0.9699Å,AH-O-H为108.4°.由此可见,在Ce–2H2O–CO2体系中基态Ce对CO2和H2O都起到了一定的影响和作用,使得CO2的键长和H2O的键长键角发生了变化.
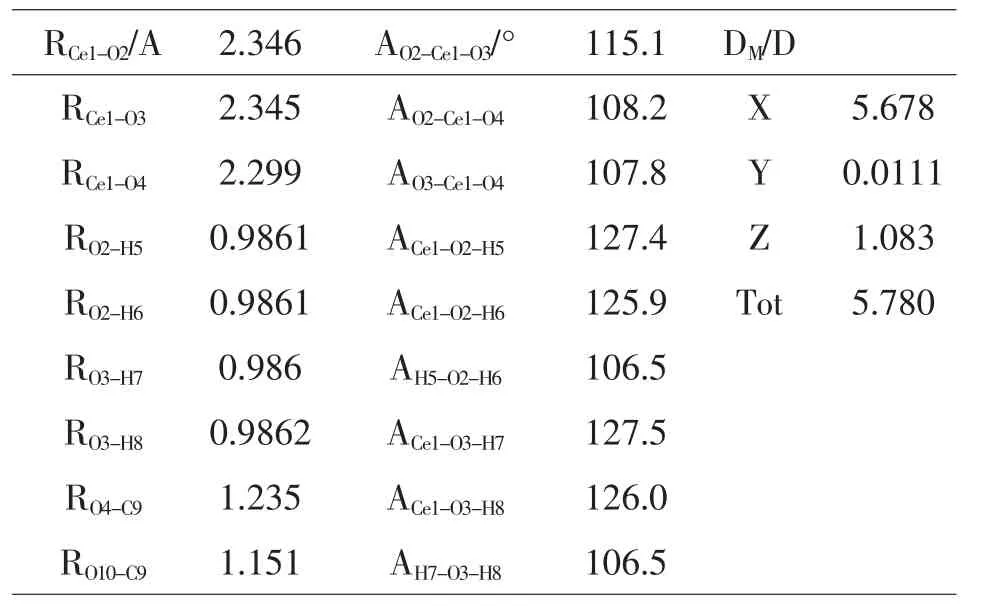
表4 -1 Ce–2H2O–CO2的几何结构参数
在Ce–2H2O–CO2–td中,C=O键是1.152Å和1.236Å,而激发态的CO2的键长为1.283Å.在Ce–2H2O–CO2–td中H–O键是0.9861Å和0.9867Å,AH-O-H是106.5°和106.6°,而激发态的H2O中的H–O键为1.280Å,AH-O-H为179.8°.由此可见,在Ce–2H2O–CO2–td体系中激发态Ce对CO2和H2O都起到了一定的影响和作用,使得CO2的键长和H2O的键长键角发生了变化.在目前研究水平上,配合物基态和激发态的结构参数变化不是很大,深层次的原因,有待进一步研究.而激发态时单个CO2分子的C=O键长比配合物中的C=O键长,水分子的键长也是这样,在激发态时,水分子的H–O为离解态,处于非键的状态.
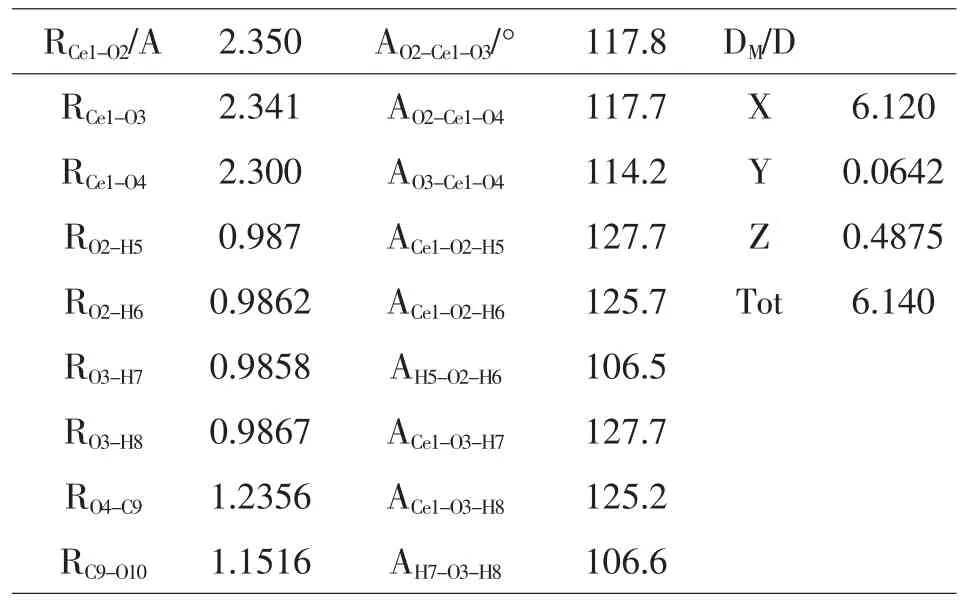
表4 -2 Ce-2H2O-CO2-td激发态的几何结构参数
由上图4-1及表4-1(Ce)和表4-2(Ce-td)我们很容易看出,在Ce–2H2O–CO2中的C=O键长为1.151Å和1.235Å,H–O键长为0.9861Å和0.986Å,AH-O-H是106.5°,在Ce–2H2O–CO2–td中,C=O键长为1.152Å和1.236Å,H–O键长为0.9861Å和0.9867Å,AH-O-H是106.5°和106.6°,在M–2H2O–CO2模型中,Ce和Ce–td对H2O和CO2都起到了一定的影响和作用,使得CO2的键长和H2O的键长键角发生了变化,但变化不大,可能是稀土离子相对复杂的f层电子结构所致,有待于进一步研究.
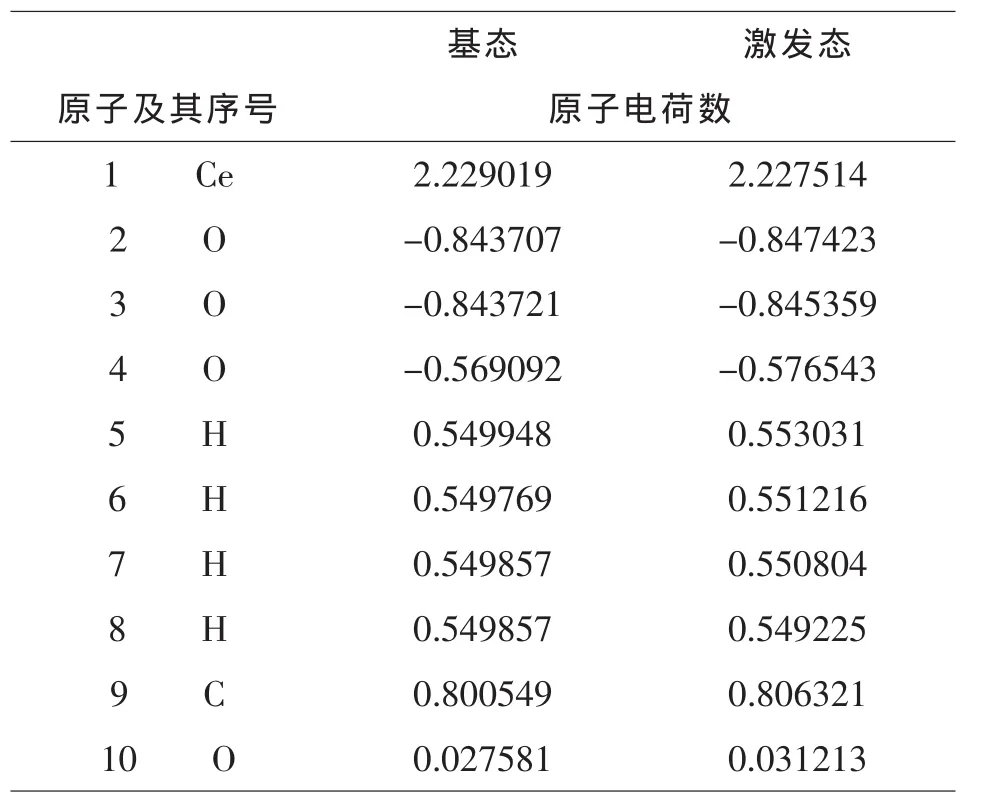
表4-3 基态和激发态基态Ce–2H2O–CO2原子电荷比较
由图4-1和表3-5,3-6可知,基态和激发态Ce与H2O和CO2分别相互作用后,每个原子的电荷数皆发生变化.由于不同状态铈离子的作用,使得配合物体系中不同原子的电荷分布不同,基态Ce的配合物中O(2)原子的电荷为:-0. 8437e-;激发态Ce的配合物中O(2)原子的电荷为:-0.8474 e-;它们相差了:-0.0037 e-,而且这些分布又不同于单分子水中的O原子的电荷,原因是铈离子的加入使得电场发生了变化.基态Ce的配合物中H(5)原子的电荷为0.5499 e-;激发态Ce的配合物中H(5)原子的电荷为:0.5530 e-;它们相差了:0.0031 e-.通过对比可以发现,激发态Ce的配合物中各个原子的电荷数变化较大.
Ce3+与反应物分子相互作用的能量分为基态和激发态两种情况研究,分析如下:
基态时的能量:
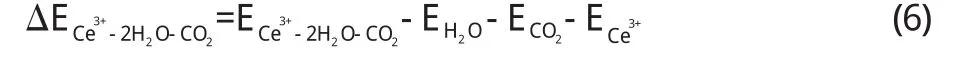

由以上(5)、(6)两个式子算出了每个部分的相互作用能,基态时的能量ΔECe3+-2H2O-CO2为–1.181×106J/mol,激发态时的能量ΔECe3+-2H2O-CO2-td为–1.065×106J/mol,激发态时的能量要大于基态时的能量,所以基态时的Ce要比激发态时Ce对反应物分子的影响大,活化作用也越大.
另外,总结2H2O-CO2-Ce-OPT和2H2O-CO2-Ce-OPT-td的自旋密度(DOS),见图4-2和图4-3.截取的能量范围是-20~0eV.
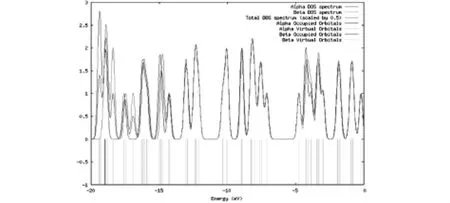
图4-2 2H2O-CO2-Ce-OPT的DOS谱图

图4-3 2H2O-CO2-Ce-OPT-td的DOS谱图
图中分别给出各种类型的α和β占据轨道和空轨道的自旋密度参数,费米能级α和β轨道分别在-19.04 eV和-18.18 eV.2H2O-CO2-Ce-OPT的α峰值出现在-19.00 eV和-15.00 eV,而β峰值集中在-15.00 eV附近和-16.00 eV及-19.00 eV处.2H2O-CO2-Ce-OPT-td的α峰出现在-13.00 eV及-19.50 eV处,β峰值主要出现在-17.50 eV及-19.00 eV处.
5 结论
通过研究铈离子与H2O和CO2分子的相互作用,采用范德华配合物模型:M–3CO2,M–2H2O–CO2(M=Ce–td,Ce (+4)和Ce(+3)),在B3LYP水平上计算完成,得到相应的优化的结构参数,和相互作用能量,通过这些结构参数和相互作用能得到以下结论:
在采用范德华配合物模型M–2H2O–CO2(M=Ce(+4)和Ce(+3))进行优化时,Ce(+4)和Ce(+3)都会对H2O和CO2分子产生影响.单个CO2分子的C=O键为1.188Å,在Ce(+4)的配合物中的C=O键是1.151Å和1.262Å,在Ce(+3)的配合物中的C=O键是1.151Å和1.235Å,Ce(+4)的配合物中的C=O键长要比Ce(+3)的配合物中的变化大,此外,对H–O键长.AH-O-H键角进行比较,比较后发现Ce(+4)的配合物中的H–O键长AH-O-H键角皆比Ce(+3)的配合物中的变化大.
Ce(+4)–2H2O–CO2的相互作用能ΔECe4+-2H2O-CO2为–2.692×106J/mol,Ce(+3)–2H2O–CO2的相互作用ΔE Ce3+-2H2O-CO2为–1.181×106J/mol,由此可以看出Ce(+4)–2H2O–CO2的相互作用能ΔE较大,ΔE越大,金属离子与反应物分子相互作用力越大,对反应物分子的影响就越大,对反应物分子的活化程度就越大,由此可见Ce(+4)要比Ce(+3)对H2O和CO2影响作用大.对基态和激发态的Ce进行了对比,对比结果为激发态Ce和基态Ce下的H2O和CO2配合物结构参数变化不是很大,深层次的原因,有待进一步研究.在能量上进行比较发现,激发态下的Ce比基态下的Ce对H2O和CO2影响作用略大,说明3价铈离子在激发态可能参与了光生电子的传输.
〔1〕Inoue T,Fujishima A,Konishi S,Honda K.Photoelectrocatalytic reduction of carbon dioxide in aqueous suspensions of semiconductor powders[J].Nature,1979, 277:637–638.
〔2〕Yui T.,Tamaki Y.,Sekizawa K.,Ishitani O.,Photocatalytic Reduction of CO 2 from Molecules to Semconductors,Top.Curr.Chem.2011,303,151-184.
〔3〕Ettedgui J.,Diskin-Posner Y.,Weiner L.,Neumann R.,Visible-Light-Driven H2 Generation from W ater and CO 2 Conversion by Using a Zw itterionic Cyclometalated Iridium(III)Complex,J.Am.Chem.Soc. 2011,133,188–190.
〔4〕Frisch M.J.,TrucksG.W.,Schlegel H.B.,Scuseria G.E., RobbM.A.,Cheeseman J.R.,Montgomery J.A.,Jr.,Vreven T.,Kudin K.N.,Burant J.C.,M illam J.M.,Iyengar S.S., Tomasi J.,Barone V.,MennucciB.,CossiM.,ScalmaniG., RegaN.,Petersson G.A.,NakatsujiH.,HadaM.,EharaM., ToyotaK.,FukudaR.,Hasegawa J.,IshidaM.,Nakajima T., Honda Y.,Kitao O.,Nakai H.,Klene M.,Li X.,J.E., H ratchian H.P.,CrossJ.B.,Bakken V.,Adamo C.,Jaram illo J.,GompertsR.,Stratmann R.E.,Yazyev O.,Austin A.J., Camm iR.,PomelliC.,Ochterski J.W.,Ayala P.Y.,Morokuma K.,Voth G.A.,Salvador P.,Dannenberg J.J.,Zakrzewski V.G.,Dapprich S.,Daniels A.D.,Strain M.C., Farkas O.,Malick D.K.,Rabuck A.D.,Raghavachari K., Foresman J.B.,O rtiz J.V.,CuiQ.,BaboulA.G.,Clifford S., Cioslow ski J.,Stefanov B.B.,Liu G.,Liashenko A.,Piskorz P.,Komarom i I.,Martin R.L.,Fox D.J.,Keith T.,Al-Laham M.A.,Peng C.Y.,Nanayakkara A.,Challacombe M., GillP.M.W.,Johnson B.,ChenW.,W ongM.W.,Gonzalez C.,Pople J.A.,Gaussian,Inc.,W allingford CT,2004.
〔5〕Lee C,YangW,Parr R G.Deve lopm ent o f the Co llesalvetti Co rre la tion-energy Formu la into a Funtional of the E lection Den fity[J].Phys Rev B, 1988,37,785-789.
〔6〕Indrakanti V P,Kub icki JD,Schobert H H.Quantum M echanicalM odeling of G round States of CO 2 Chem isorbed on Anatase(001),(101)and(110)Surfaces[J].Energy&Fue ls,2008,22:2611-2618.
〔7〕Hehrew J,Ditchfield R,Pople J A.,Se lf-Consistent Molecular O rbital M ethods.XII.Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of O rganic Molecules[J].J.Chem Phys,1972, 56:2257-2261.
〔8〕Hay P J,W adtW R.,Ab in itio effective core potentials for molecular calculations for the transition metal atoms Sc to Hg,J.Chem.Phy s.1985,82:270-283;Hay P J,W adtW R.Ab initio effective core potentia ls formo lecular calculations,poten tialsformain group elementsNa to Bi,J.Chem. Phys.1985,82:284-298;Hay P J,W adtW R.,Ab in itio effective core potentials formolecular calculations,potentials for K to Au including the outermost core orbitals,JChem Phys,1985,82:299-310.
O643
A
1673-260X(2014)07-0009-05
