一种通用型piggyBac转座子诱导细胞永生化载体的构建及其基本功能验证
2014-06-24黄惠胡广东康健卿素珠张涌
黄惠,胡广东,康健,卿素珠,张涌
1 西北农林科技大学动物医学院, 陕西 杨凌 712100
2 西北农林科技大学 农业部动物生物技术重点实验室, 陕西 杨凌 712100
动物及兽医生物技术
一种通用型piggyBac转座子诱导细胞永生化载体的构建及其基本功能验证
黄惠1,2,胡广东1,2,康健1,2,卿素珠1,张涌1,2
1 西北农林科技大学动物医学院, 陕西 杨凌 712100
2 西北农林科技大学 农业部动物生物技术重点实验室, 陕西 杨凌 712100
为了构建一种高效、通用的细胞永生化载体pTP-hTERT,通过人工合成、PCR、酶切连接等方法,对传统piggyBac (PB) 转座子系统进行改造。改造后的载体含有转座必需元件、PB转座酶 (PBase) 表达框、共表达筛选元件和人端粒逆转录酶 (hTERT) 基因表达框。其中筛选元件中绿色荧光蛋白 (EGFP)基因和嘌呤霉素抗性 (Puror) 基因以猪捷申病毒2A自我剪切肽相连,以实现共表达。为验证载体元件功能,使用该载体转染HEK 293细胞,并对筛选出的阳性细胞进行RT-PCR、Western blotting (WB)、热不对称PCR (Tail-PCR) 和细胞克隆亚甲蓝染色与统计分析。载体测序鉴定与细胞培养结果表明,通用型永生化载体 pTP-hTERT构建成功,转染HEK293细胞后能筛选出抗嘌呤霉素细胞单克隆;WB结果显示P2A可高效切割EGFP和Puror融合蛋白,证明筛选标记基因功能正常;Tail-PCR结果表明该载体以转座整合插入宿主基因组;亚甲蓝染色统计结果显示由pTP-hTERT引发的细胞阳性克隆数与对照组相比显著提高 (P<0.01)。PB转座子永生化载体pTP-hTERT的构建为永生化细胞系的建立提供了工具,同时也为其他真核载体的构建和改造提供了参考。
piggyBac (PB) 转座子,P2A,融合蛋白,hTERT
转座子 (Transposon) 是一类能在宿主基因组中变更插入位置的可移动遗传因子,其变更插入位置的过程被称为转座 (Transposition)。piggyBac (PB) 转座子是病毒遗传学家Fraser等最初发现并构建二元转座系统证明其不仅仅在一个动物宿主中具有转座活性[1]。自中国学者证明该转座子可在哺乳动物细胞中高效转座以来[2],PB转座系统逐渐作为一种高效基因传递工具被应用于各种哺乳动物基因工程试验中,如反向遗传筛选[3]、转基因动物生产[4-5]、iPS诱导[6-7]、RNA干扰[8]、基因治疗[9]以及诱导突变[10-11]等。
PB转座的发生是由PB转座酶 (PB transposase,PBase) 介导的,实际应用过程中,PB转座系统是由一个供体质粒和一个辅助质粒组成的二元系统[12]。供体质粒包含转座发生所必需的5′TR和3′TR核苷酸序列[13],并承担携带筛选报告基因表达框和外源基因的功能,辅助质粒主要包含一个PBase表达框,供体质粒和辅助质粒按比例共转染细胞后,在PBase的作用下,供体质粒中的转座元件特异性插入宿主基因组TTAA位点[14-15]。有报道表明PB转座系统携带100 kb大小的外源基因依然具有较高活性[16],因而该系统在实现大片段基因或多基因在哺乳动物细胞中的表达具有较广阔的应用前景。
本研究对传统PB转座子二元质粒系统进行改造,将供体质粒和辅助质粒整合组成单质粒PB转座系统,利用猪捷申病毒2A自剪肽(Porcine teschovirus-1 2A peptide, P2A)连接绿色荧光蛋白 (Enhanced green fluorescent protein,EGFP) 和嘌呤霉素抗性 (Puromycinresistance,Puror) 基因组成筛选标记基因表达框,并在该表达框的两端引入Loxp序列。同时,插入由CMV启动子介导的人端粒酶逆转录酶表达框,最终构建成功由PB转座子介导hTERT表达的永生化载体,将该载体转染HEK293细胞后,对载体的基本特性进行鉴定。
1 材料与方法
本试验于2012年6月至2013年8月在西北农林科技大学农业部直属动物生物技术国家重点实验室进行。
1.1 材料
1.1.1 质粒、菌株和细胞
质粒pMD18-T Simple、pMD18-T购自宝生物工程 (大连) 有限公司;pEGFP-C1、pEF1α-Tet3G购自美国Clontech Laboratories;pCDH-MCS-T2A-Puror-MSCV购自于美国System Biosciences SBI公司;质粒pCI-neohTERT由西北农林科技大学动物医学院张彦明教授惠赠;大肠杆菌DH5α感受态购自于全式金 (北京) 生物技术有限公司;pBNW-TP2质粒由本实验室构建并保存;HEK293细胞系购自中国科学院细胞库。
1.1.2 工具酶和试剂
内切酶均购自NEB公司。T4 DNA连接酶、PrimeSTAR DNA聚合酶、rTaq DNA聚合酶、染色体步移试剂盒购自宝生物工程 (大连) 有限公司。Blue PlusProteinⅡmarker、GFP单克隆抗体、羊抗兔二抗、Pfu DNA聚合酶购自全式金 (北京) 生物技术有限公司;质粒提取试剂盒购于生工生物工程 (上海) 股份有限公司;凝胶回收试剂盒均购自爱思进生物技术 (杭州)有限公司;PureYield™ PlasmidMaxiprep System购自普洛麦格 (北京) 生物技术有限公司;嘌呤霉素购自Sigma公司;胎牛血清FBS购自Hyclone公司;DMEM培养基、Lipofectamine 2000购自Invitrogen公司。
1.2 永生化载体pTP-hTERT构建
1.2.1 单质粒转座子载体pTP的构建
PB转座子基本骨架载体pBNW-TP2由生物公司合成,包含PB转座子5′TR (313 bp) 和3′TR (235 bp) 必需核苷酸序列,一个由TK启动子调节的PBase表达框,一个由12个稀有酶切位点组成的MCS及2个拷贝的cHS4绝缘子序列。
筛选标记基因表达框的构建,参考冷泉港实验室重叠延伸PCR实验策略[17],分别以pEGFP-C1、pCDH-MCS-T2A-Puror-MSCV为模板,用PrimeSTAR DNA聚合酶扩增EGFP-P2A和P2A-Puror序列 (引物序列见表1),凝胶回收PCR产物。按以下比例制备重叠延伸PCR反应体系:5 μL 10×Pfu缓冲液、0.7 μL Pfu DNA聚合酶、4 μL dNTPs、1 μL EGFP片段、1 μL Puror片段、33 μL ddH2O。反应条件:94 ℃预变性5 min;先94 ℃变性30 s、69 ℃退火30 s、72 ℃延伸1 min,3个循环,每个循环退火温度降低2 ℃,再94 ℃变性30 s、60 ℃退火30 s、72 ℃延伸2 min,33个循环。反应过程中3个循环后暂停反应,向体系中加入EGFP上游引物和Puror下游引物 (引物序列见表1) 各1 μL、0.3 μL Pfu DNA聚合酶,继续反应直至结束。将得到的EGFP-P2A-Puror片段,NheⅠ、KpnⅠ酶切连入pEGFP-C1载体。用PrimeSTAR DNA聚合酶扩增pEF1α-Tet3G中EF1α启动子序列 (引物序列见表1),经AseⅠ、NheⅠ酶切连接EGFP-P2A-Puror,完成筛选表达框EF1α-EGFP-P2A-Puror-polyA的构建,所有扩增片段必须经过测序。
设计含有LoxP的引物 (引物序列见表1),以高保真PrimeSTAR DNA聚合酶扩筛选表达框,得到两端带有LoxP序列的筛选元件。再经SpeⅠ、SalⅠ酶切连至pBNW-TP2载体中,构成了带有筛选标记的单质粒PB转座子载体pTP。
1.2.2 hTERT表达框的的引入
构建含有SpeⅠ、AscⅠ酶切位点、多克隆位点 (MCS)和polyA的质粒pMD18-T simple-MCS-polyA (引物序列见表1)。将pCl-neo-hTERT中CMV-hTERT,酶切连入MCS,得到完整的hTERT表达框CMV-hTERT-polyA。通过引入的SpeⅠ和AscⅠ酶切位点表达框连入一元转座子载体pTP,构成永生化载体pTP-hTERT。

表1 实验所用引物序列Table 1 Primers used in experiments
1.3 载体功能验证
1.3.1 HEK293细胞转染及筛选
用10% FBS的DMEM培养HEK293细胞。转染试验严格按照Lipofectamine 2000操作说明书要求进行。转染前24 h,以无双抗培养基接种HEK293细胞于6孔板,使转染时细胞融合度为60%−80%,每孔加入Lipofectamine 2000 10 μL、质粒pTP-hTERT 4 μg,5 h后换液,24 h后观察。
筛选阳性单克隆前,应先确定最佳筛选浓度。具体操作如下:培养带转染细胞,当细胞融合度达到70%−80%,更换为嘌呤霉素浓度分别为0、0.3、0.6、0.8、1、1.5、2、3、5 μg/mL的新鲜无双抗培养基。7 d内致所有细胞死亡的最小浓度即为最佳筛选浓度。
以Lipofectamine 2000介导pTP-hTERT转染HEK293细胞,24 h后接种于90 mm皿,以含有最佳筛选浓度嘌呤霉素的培养基筛选阳性克隆,阴性对照完全死亡后,嘌呤霉素浓度减半,扩大培养。
1.3.2 RT-PCR检测hTERT转录
按照标准操作对扩大培养的单克隆细胞RNA提取及反转录PCR (引物序列见表1)。
1.3.3 Western blotting检测P2A剪切效率
以pEGFP-C1为阳性对照,正常细胞为阴性对照。裂解 (未) 转染pTP-hTERT (pEGFP-C1)后48 h的HEK293细胞,提取蛋白,使用12% SDS-PAGE蛋白胶、Blue PlusProteinⅡmarker、兔源GFP单克隆抗体,以GAPDH内参进行Western blotting。
1.3.4 基因组转座位点检测
用pTP-hTERT转染HEK293细胞,经嘌呤霉素筛选出9株细胞克隆,同时设立转pEGFP-C1质粒的细胞为阳性对照组,并筛选出9株对照组细胞克隆。以载体上转座元件TR序列为模板设计引物,以细胞克隆基因组为模板进行Tail-PCR,扩增结束后利用琼脂糖凝胶电泳鉴定,特异性DNA片段胶回收后克隆至pMD18-T载体,筛选阳性重组子送至南京金斯瑞生物公司测序,测序结果登陆美国加州大学基因组生物信息学网站 (http://genome.ucsc.edu/)进行分析,确定整合入基因组的转座子的侧翼序列[18],及染色体整合位点具体信息。Tail-PCR试验步骤参照染色体步移试剂盒 (TAKARA)操作说明书 (引物序列见表1)。
1.3.5 转座效率检测
将pPB-hTERT和本实验构建的pBNW-TP1 (即随机整合质粒) 均以0.25 pmol的量分别转染约1×106个HEK 293细胞,细胞铺板至90 mm平皿,嘌呤霉素筛选出单克隆后,用含4%的多聚甲醛固定,0.2%亚甲蓝溶液染色30 min,去离子水清洗后,统计细胞克隆数,本操作同等条件下重复3次,统计结果以平均数±标准误表示,利用非配对t检验 (Student’s t-test) 对细胞克隆数进行统计学分析。
2 结果与分析
2.1 永生化载体pTP-hTERT构建
2.1.1 单质粒转座子载体pTP的鉴定结果
全载体图谱如图1所示。
PB转座子骨架载体pBNW-TP2的鉴定,用SpeⅠ单酶切待改造pBNW-TP2质粒,经凝胶电泳鉴定为6.3 kb (图2),大小与预期相符,可用于进一步的载体改造。

图1 质粒pTP-hTERT图谱Fig. 1 Map of pTP-hTERT.

图2 质粒pBNW-TP2的酶切鉴定Fig. 2 Identification of primary vector pBNW-TP2. M: Trans 15K DNA marker; 1: pBNW-TP2 digested with Spe.Ⅰ
pTP载体初步鉴定,PCR扩增EGFP-P2A和 P2A-Puror序列,凝胶电泳鉴定大小为838 bp和687 bp,重叠延伸PCR反应产物EGFPP2A-Puror凝胶电泳鉴定大小为1 489 bp (图3),EF1α序列PCR产物凝胶电泳鉴定为1 317 bp (图4),皆与预期相符。以PCR引物形式引入酶切位点及LoxP序列后得到的筛选元件LoxP-EF1α-EGFP-P2A-Puror-polyA-LoxP经凝胶电泳鉴定大小为3 150 bp (图5),符合预期大小。一元转座子载体pTP经过SpeⅠ和SalⅠ酶切得到3 142 bp的筛选元件和6.3 kb的转座子骨架 (图6),与预期结果一致。另外,所有PCR片段测序结果与原序列比对的结果显示100%同源无突变,说明克隆得到改造后转座子载体pTP正确。
2.1.2 pTP-hTERT载体酶切鉴定
hTERT表达框中只有引入的插入位点MCS-polyA为PCR扩增所得,产物凝胶电泳鉴定为326 bp (图7),测序结果显示无突变。最终载体pTP-hTERT经过SpeⅠ和AscⅠ酶切得到4 811 bp和9.6 kb的片段 (图8),即hTERT表达框和线性一元转座子载体pTP,与预期结果一致,证明PB转座子介导的人端粒逆转录酶催化亚基表达载体pTP-hTERT构建成功。
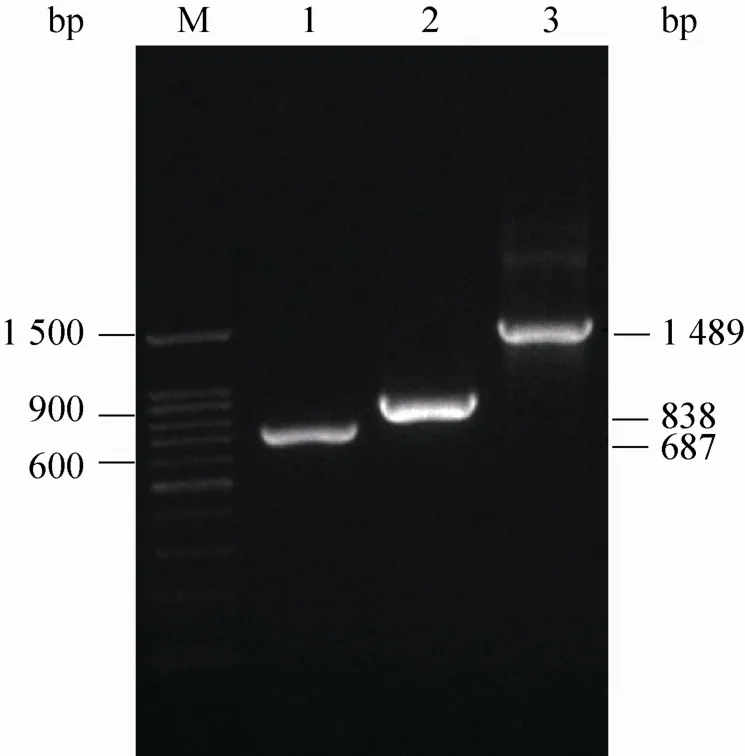
图3 重叠延伸PCR产物Fig. 3 The products of overlap PCR. M: Trans 100 bp DNA marker; 1: P2A-Puror; 2: EGFP-P2A; 3: EGFP-P2A-Puror.

图4 EF1α启动子序列的PCR检测结果Fig. 4 PCR result of EF1α promoter. M: Trans 2K Plus DNA marker; 1: EF1α amplified by PCR.

图5 筛选元件PCR扩增结果Fig. 5 PCR result of selectable-reporter. M: Trans2K Plus DNA marker; 1, 2: selectable element LoxP-EF1α-EGFP-P2A-Puror-polyA-LoxP amplified by PCR.
2.2 pTP-hTERT载体在HEK293细胞中的功能验证结果
2.2.1 HEK293细胞瞬时转染及筛选结果
待转染的HEK293细胞,转染pTP-hTERT 48 h后的镜下观察结果 (图9A,B)。HEK293细胞嘌呤霉素最佳筛选浓度测定结果为1 μg/mL。转染后的HEK293细胞,经过6 d筛选后,得到细胞形态正常的阳性克隆,阳性细胞挑取并扩大培养 (图9C,D),已进行后续检测。

图6 质粒pTP的酶切鉴定Fig. 6 Identification of modified vector pTP by double digestion. M: GenStar 1 kb DNA marker; 1: pTP digested with SpeⅠand SalⅠ.
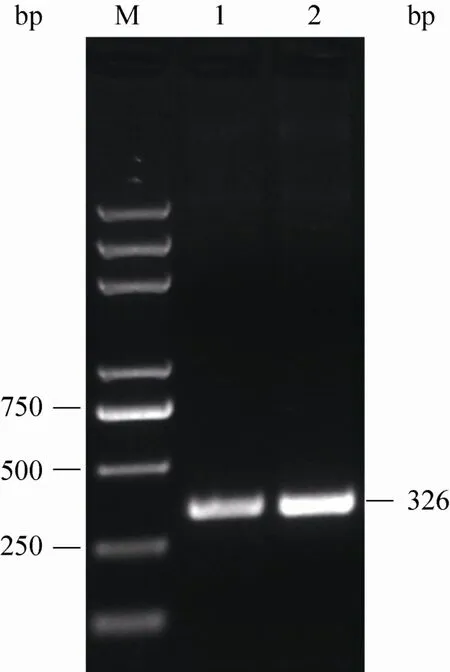
图7 MCS PCR扩增结果Fig. 7 PCR result of MCS. M: Trans 2K Plus DNA marker; 1, 2: MCS-polyA amplified by PCR.
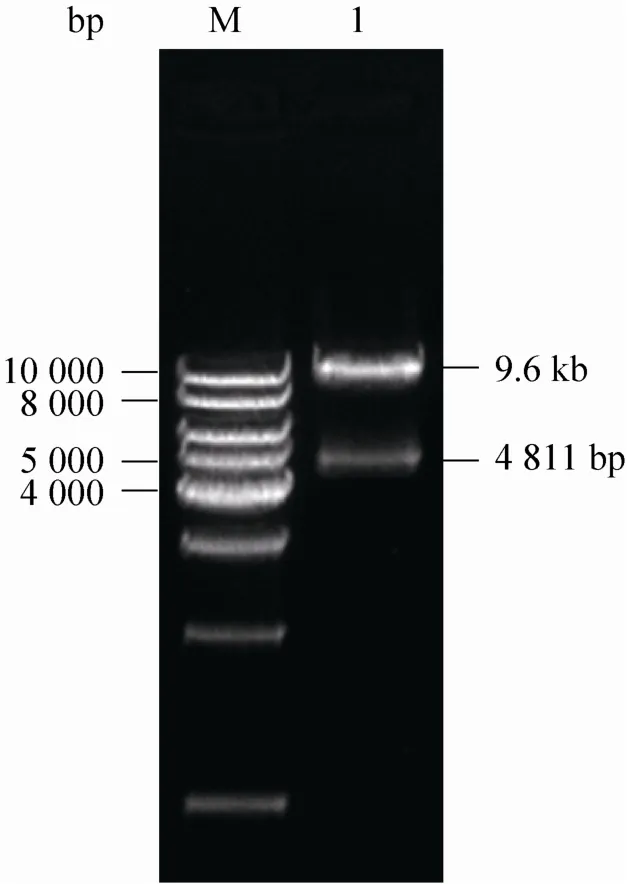
图8 质粒pTP-hTERT的酶切鉴定Fig. 8 Identification of pTP-hTERT by double digestion. M: GenStar1kb DNA marker; 1: pTP-hTERT digested with SpeⅠand AscⅠ.

图9 瞬时转染HEK293细胞及单克隆细胞扩大培养 (100倍)Fig. 9 HEK293 cells transfected with pTP-hTERT and selected monoclonal cells. (A and B) HEK293 cells 48 hours after transfection (magnification×100). (C and D) Puromycin-resisted monoclonal cells (magnification×100).
2.2.2 RT-PCR检测hTERT转录
对所提取的总RNA反转录后,用hTERT检测引物进行PCR,经凝胶电泳检测得到553 bp片段,与预期大小一致 (图10)。
2.2.3 Western blotting检测P2A剪切效率
pEGFP-C1转染细胞为对照,正常细胞为阴性对照,GAPDH为内参。EGFP蛋白条带约位于30 kDa,EGFP-Puror融合蛋白位于55 kDa左右,样品中融合蛋白条带细而EGFP条带清晰(图11),表明样品P2A连接的融合蛋白大多被正确剪切,即载体中的P2A自我剪切肽整合入细胞后能正常发挥其剪切功能。2.2.4 基因组转座位点检测
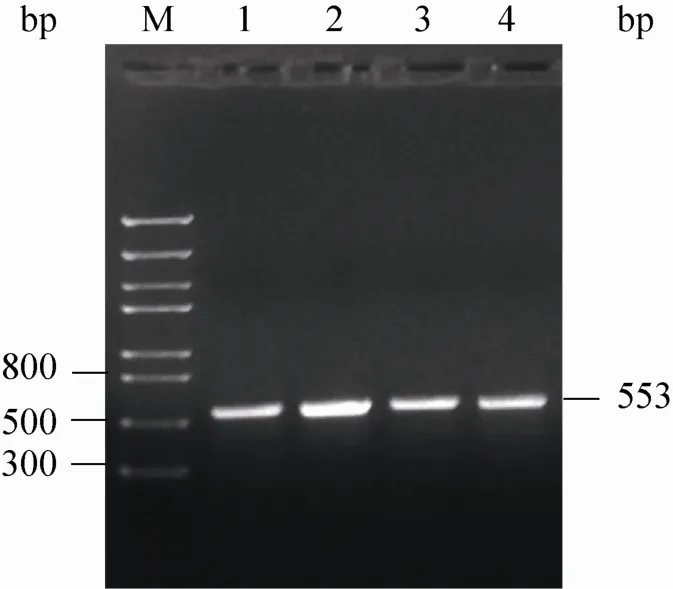
图10 单克隆细胞hTERT基因RT-PCR鉴定Fig. 10 RT-PCR analysis of hTERT gene in cell clones. M: Trans 5K DNA marker; 1−4: cDNA of hTERT fragment in four independent cell clones.

图11 Western blotting检测P2A剪切功能Fig. 11 Validation of P2A by Western blotting analysis.
对试验所获得的基因组转座子侧翼序列进行比对分析后,发现9个实验组样品整合位点侧翼序列可定位于人2号、4号、6号、12号、13号、 21号染色体 (具体整合位点信息见表2)。该结果表明,构建的pTP-hTERT转座子可转座整合入宿主细胞基因组。
2.2.5 细胞克隆计数统计结果
三次细胞克隆计数结果经非配对t检验(Student’s t-test) 分析后,结果如图12所示。结果显示不含PB转座酶的质粒pBNW-TP1转染细胞后得到的细胞克隆数远低于转染pTB-hTERT得到的的细胞克隆数,两者之间差异极显著 (P<0.01)。

图12 HEK293 细胞中PB转座效率统计结果Fig. 12 The statistical result of PB transposon integration efficiency in HEK293 cell.

表2 HEK293 细胞中PB转座子整合位点侧翼序列及具体信息Table 2 Flanking sequences of piggyBac integration sites in HEK 293 cells
3 讨论
有研究显示,PB转座子在真核细胞中的整合效率是传统随机整合载体的8倍以上[2],并且单质粒PB转座子载体相较于传统双质粒转座子系统转座效率更高[19]。本研究将二元PB转座子改造为单质粒载体介导外源基因整合,并对其筛选报告元件进行了优化。对改造后的单质粒转座载体pTP-hTERT和传统二元转座供体质粒载体进行转座效率检测,统计分析结果显示单质粒转座载体转染细胞后产生的阳性克隆数远高于不加转座酶的供体质粒 (P<0.01),表明构建的载体能够高效整合入基因组。另外,对转染pTP-hTERT筛选得到的多株阳性细胞基因组进行转座子整合位点侧翼DNA序列检测,结果显示基因组整合位点TTAA两侧并无转座元件外侧的载体序列,证实该质粒的整合方式为转座整合而非随机整合。实验证明载体pTP-hTERT能够携带外源基因进行高效转座。
为了实现多个基因同时在动物细胞中进行表达,通常需要不同启动子分别介导目的基因的转录。然而多个表达框的引入不仅存在启动子之间的相互干扰,并且容易使随机整合载体容量超载。内部核糖体进入序列 (IRES) 可实现多顺反子中多个编码基因的单独表达,但在实际操作过程中,其上下游基因表达不平衡以及自身结构较大等问题使其应用严重受限[20]。2A自剪肽是存在于多种病毒中的短肽链,能够介导融合蛋白自我剪切[21]。由于其结构短小,介导的蛋白剪切效率较高,因而被越来越多应用于多顺反子载体构建中。有研究表明,猪捷申病毒2A (Porcine teschovirus-1 2A peptide,P2A)相对于其他病毒2A剪切效率更高[22]。因此,pTP-hTERT中使用P2A介导EGFP和Puror融合蛋白剪切,经Western blotting验证,P2A可有效剪切EGFP-Puror融合蛋白,保证了筛选基因的正常功能,这为多顺反子真核表达载体的构建提供了参考价值。
抗性筛选基因的长期存在可破坏整合位点附近正常功能基因的表达[23],为了解决该问题,设计引入Cre/LoxP系统来最终达到切除筛选标记基因的目的。通过细胞转染和筛选获取阳性克隆细胞后,利用原核表达Cre重组酶来实现筛选标记基因的切除[24],从而消除其对宿主细胞功能基因表达的负面影响。
本实验选用hTERT诱导细胞永生化能最大限度保留正常细胞特性,是大多数细胞逃避衰老的最安全有效的手段[25-26]。由于hTERT编码区序列较长,本实验利用PB转座系统来携带该基因,既发挥了PB转座子高效整合的特点,又利用了其负载容量大的优势。
综上所述,本实验设计构建了一种通用的PB转座子介导的单质粒hTERT表达载体,并在HEK 293细胞中对该载体基本功能进行验证。结果表明载体筛选基因能正确表达及剪切,PB转座子元件携带目的基因可在宿主细胞基因组上定点插入TTAA位点,发生PB转座,即证明具有PB转座活性的载体构建成功。
REFERENCES
[1] Fraser MJ, Cary L, Boonvisudhi K, et al. Assay for movement of Lepidopteran transposon IFP2 in insect cells using a baculovirus genome as a target DNA. Virology, 1995, 211(2): 397–407.
[2] Ding S, Wu X, Li G, et al. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell, 2005, 122(3): 473–483.
[3] Rad R, Rad L, Wang W, et al. PiggyBac transposonmutagenesis: a tool for cancer gene discovery in mice. Science, 2010, 330(6007): 1104–1107.
[4] Bai DP, Yang MM, Chen YL. PiggyBac transposon-mediated gene transfer in Cashmere goat fetal fibroblast cells. Biosci Biotechnol Biochem, 2012, 76(5): 933–937.
[5] Park TS, Han JY. PiggyBac transposition into primordial germ cells is an efficient tool for transgenesis in chickens. Proc Natl Acad Sci USA, 2012, 109(24): 9337–9341.
[6] Woltjen K, Michael IP, Mohseni P, et al. PiggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature, 2009, 458(7239): 766–770.
[7] Desponts C, Ding S. Using small molecules to improve generation of induced pluripotent stem cells from somatic cells. Methods Mol Biol, 2010, 636: 207–218.
[8] Zhou F, Liang S, Chen AH, et al. A transgenic Marc-145 cell line of piggyBac transposon-derived targeting shRNA interference against porcine reproductive and respiratory syndrome virus. Vet Res Commun, 2012, 36(2): 99–105.
[9] Feschotte C. The piggyBac transposon holds promise for human gene therapy. Proc Natl Acad Sci USA, 2006, 103(41): 14981–14982.
[10] Kim A, Pyykko I. Size matters: versatile use of PiggyBac transposons as a genetic manipulation tool. Mol Cell Biochem, 2011, 354(1/2): 301–309. [11] Yusa K, Zhou L, Li MA, et al. A hyperactive piggyBac transposase for mammalian applications. Proc Natl Acad Sci USA, 2011, 108(4): 1531–1536.
[12] Wang W, Lin C, Lu D, et al. Chromosomal transposition of PiggyBac in mouse embryonic stem cells. Proc Natl Acad Sci USA, 2008, 105(27): 9290–9295.
[13] Li X, Harrell RA, Handler AM, et al. PiggyBac internal sequences are necessary for efficient transformation of target genomes. Insect Mol Biol, 2005, 14(1): 17–30.
[14] Elick TA, Bauser CA, Fraser MJ. Excision of the piggyBac transposable element in vitro is a precise event that is enhanced by the expression of its encoded transposase. Genetica, 1996, 98(1): 33–41.
[15] Liang Q, Kong J, Stalker J, et al. Chromosomal mobilization and reintegration of Sleeping Beauty and PiggyBac transposons. Genesis, 2009, 47(6): 404–408.
[16] Li MA, Turner DJ, Ning Z, et al. Mobilization of giant piggyBac transposons in the mouse genome. Nucleic Acids Res, 2011, 39(22): e148.
[17] Szymczak-Workman AL, Vignali KM, Vignali DA. Vignali. Design and construction of 2A peptide-linked multicistronic vectors. Cold Spring Harb Protoc, 2012(2): 199–204.
[18] Liu YG, Chen Y. High-efficiency thermal asymmetric interlaced PCR for amplification of unknown flanking sequences. Biotechniques, 2007, 43(5): 649–656.
[19] Urschitz J, Kawasumi M, Owens J, et al. Helper-independent piggyBac plasmids for gene delivery approaches: strategies for avoiding potential genotoxic effects. Proc Natl Acad Sci USA, 2010, 107(18): 8117–8122.
[20] Liu BS, Liu XY, Qian C. An efficient tool for the construction of multiple-cistronic vectors: FMDV 2A. Chin J Biotech, 2007, 23(5): 765–769 (in Chinese).
刘必胜, 刘新垣, 钱程. 构建多顺反子表达载体的有效工具-FMDV 2A. 生物工程学报, 2007, 23(5): 765–769.
[21] Palmenberg AC, Parks GD, Hall DJ, et al. Proteolytic processing of the cardioviral P2 region: primary 2A/2B cleavage in clone-derived precursors. Virology, 1992, 190(2): 754–762.
[22] Kim JH, Lee SR, Li LH, et al. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS ONE, 2011, 6(4): e18556.
[23] Pham CT, MacIvor DM, Hug BA, et al. Long-range disruption of gene expression by a selectable marker cassette. Proc Natl Acad Sci USA, 1996, 93(23): 13090–13095.
[24] Yu Y, Wang Y, Tong Q, et al. A site-specific recombinase-based method to produce antibiotic selectable marker free transgenic cattle. PLoS ONE, 2013, 8(5): e62457.
[25] Toouli CD, Huschtscha LI, Neumann AA, et al. Comparison of human mammary epithelial cells immortalized by simian virus 40 T-Antigen or by the telomerase catalytic subunit. Oncogene, 2002, 21(1): 128–139.
[26] Gudjonsson T, Villadsen R, Rønnov-Jessen L, et al. Immortalization protocols used in cell culture models of human breast morphogenesis. Cell Mol Life Sci, 2004, 61(19/20): 2523–2534.
(本文责编 陈宏宇)
Construction of a general piggyBac transposon inducible cell immortalization vector and verification of its basic properties
Hui Huang1,2, Guangdong Hu1,2, Jian Kang1,2, Suzhu Qing1, and Yong Zhang1,2
1 College of Veterinary Medicine, Northwest A&F University, Yangling 712100, Shaanxi, China
2 Key Laboratory of Animal Biotechnology, Ministry of Agriculture, Northwest A&F University, Yangling 712100, Shaanxi, China
In order to construct generally efficient cell immortalization vector, pTP-hTERT, we modified the traditional piggyBac (PB) transposon using artificial synthesis, PCR and enzyme digestion. The modified vector contained the necessary transposon elements, a PB transposase expression cassette, a co-expression selectable element and a human telomerase reverse transcriptase (hTERT) expression cassette. The co-expression selectable element had two markers, enhanced green fluorescent protein (EGFP) gene and puromycin-resistance (Puror) gene, linked by porcine teschovirus-1 2A peptide (P2A). To validate the functionality of vector elements, we transfected pTP-hTERT into HEK293 cell, selected the positive cell clones and then conducted RT-PCR, Western blotting (WB) and Tail-PCR, methylene blue staining and statistic analysis on selected cells. The results of sequencing and cell culture show that the pTP-hTERT was constructed successfully and the positive cell could be selected by puromycin. The WB results, P2A cutting EGFP and Purorfusion protein with high efficiency, reflected the selectable element worked. The sequencing result of Tail-PCR confirmed the vector integrated into the genome through transposition. The results of methylene blue staining and statistic analysis indicated the clone of positive cells triggered by pTP-hTERT significantly increased (P<0.01) compared with control group. The construction of pTP-hTERT provides an efficient tool for establishing immortalized cell lines and a demonstration for building other eukaryotic plasmids.
piggyBac (PB) transposon, P2A, fusion protein, hTERT
October 13, 2013; Accepted: January 7, 2014
Suzhu Qing. Tel: +86-29-87092438; E-mail: suzhuqing@163.com
黄惠, 胡广东, 康健, 等. 一种通用型piggyBac转座子诱导细胞永生化载体的构建及其基本功能验证. 生物工程学报,2014, 30(8): 1182–1192.
Huang H, Hu GD, Kang J, et al. Construction of a general piggyBac transposon inducible cell immortalization vector andverification of its basic properties. Chin J Biotech, 2014, 30(8): 1182–1192.
Supported by: National Major Project for Production of Transgenic Breeding (No. 2013ZX08007-004), National High Technology Research and Development Program of China (863 Program) (No. 2011AA100303).
Yong Zhang. Tel: +86-29-87080085; E-mail: yongzhang19562012@gmail.com
国家转基因重大项目专项 (No. 2013ZX08007-004),国家高技术研究发展计划 (863计划) (No. 2011AA100303) 资助。
