凝胶渗透色谱-高效液相色谱-线性离子阱质谱法测定膳食样品中的氨基甲酸酯类农药残留
2014-05-08赵云峰吴永宁
杨 欣, 李 鹏, 苗 虹, 赵云峰, 吴永宁
(1.国家食品安全风险评估中心,北京100021;2.卫生部食品安全风险评估重点实验室,北京100021)
氨基甲酸酯类农药是继有机磷类农药后发展较为迅速的一类高效、广谱杀虫剂。20世纪70年代以来,由于有机氯类农药相继被禁用或者限制使用,以及抗有机磷类农药的昆虫品种的日益增多,氨基甲酸酯类农药被广泛应用于粮食、蔬菜和水果等各种农作物的生产[1],是中国目前使用量较大的杀虫剂之一。常见的氨基甲酸酯类农药有涕灭威、克百威、甲萘威、灭多威、残杀威和灭害威等。
残留在食品中的各种农药(特别是高毒农药)对人体健康的风险评估是许多国家政府机构和国际组织用作农药登记评审和农药安全性评价的重要依据[2,3]。因此,开展农药膳食风险评估研究对合理使用农药、保护公众身体健康和保证食品安全有着非常重要的意义[4,5]。氨基甲酸酯类农药降解产物通常具有与母体化合物相同甚至更强的活性。比如,涕灭威亚砜具有比涕灭威和涕灭威砜更强的胆碱酯酶抑制作用,因此,世界卫生组织/联合国粮食与农业组织/农药残留联席会议(World Health Organization/Food and Agriculture Organization/Joint Meeting on Pesticide Residues,WHO/FAO/JMPR)规定检测和评估某些农药时需同时考虑降解产物的活性影响[6]。例如,在评估涕灭威时,定义涕灭威为涕灭威及其砜、亚砜之和,以涕灭威表示;克百威为克百威和3-羟基克百威之和,以克百威表示,等等。
膳食样品不同于一般食物样品,是经烹调、加工、聚类、混合制备成的样品,因此样品不仅油脂含量高,基质更复杂,而且样品经聚类后混合相当于被进一步稀释,其中的污染物含量很低,对样品的前处理和检测方法的灵敏度和准确度要求更高。目前文献报道的氨基甲酸酯类农药残留分析方法主要有:气相色谱法[7]、气相色谱-质谱联用法[8]、液相色谱法和液相色谱-质谱联用法[9-12]、酶联免疫法[13],样品基质有尿液[9]、水[10,11]、水果[12]、蔬菜[13]和茶叶[14]等。
离子阱质谱在一次进样过程中可以得到二级和三级子离子的全扫描质谱图,因此在对目标物做定性确证分析时,获得的化合物结构信息比四极杆质谱多。同时,利用三级子离子定量可以提高信噪比,降低复杂基质的干扰。为评估中国人群氨基甲酸酯类农药膳食暴露,我们以JMPR农药残留暴露评估需同时监测农药母体和降解产物的规定为依据,按照中国总膳食研究(Chinese TDS)食物样品分类原则[15],挑选基质较为复杂的蔬菜类和肉类样品为样品研究基质,利用线性离子阱三级质谱灵敏度高、特异性强,能有效排除基质干扰的特点,建立了同时测定多种氨基甲酸酯类农药及其降解产物(涕灭威、涕灭威砜、涕灭威亚砜、甲奈威、克百威、3-羟基克百威、丁硫克百威、灭多威、硫双灭多威、甲硫威、残杀威、灭害威、乙硫苯威、乙硫苯威砜、乙硫苯威亚砜)的高效液相色谱-串联线性离子阱质谱法(HPLCLIT-MS3)。该方法灵敏、准确,能够满足氨基甲酸酯类农药膳食评估的要求。
1 实验部分
1.1 仪器与试剂
LTQ液相色谱-质谱联用仪(美国Thermo Fisher Scientific公司):配有电喷雾离子源(ESI)及Xcalibur 2.5数据处理系统;AccuPrep MPSTM凝胶渗透色谱仪(美国J2 Scientific公司);Sigma3K-15离心机(美国Sigma公司):配有12156型转子;Milli-Q超纯水器(美国Millipore公司)。甲醇、乙腈和丙酮为色谱纯(美国Fisher公司);环己烷、乙酸乙酯为色谱纯(美国J.T.Baker公司);甲酸、甲酸铵为色谱纯(美国Tedia公司);实验用水为经Milli-Q超纯水器过滤的超纯水。
标准品:涕灭威、涕灭威砜、涕灭威亚砜、甲奈威、克百威、3-羟基克百威、丁硫克百威、灭多威、硫双灭多威、甲硫威、残杀威、灭害威、乙硫苯威、乙硫苯威砜、乙硫苯威亚砜(纯度均≥96.5%,德国Dr.Ehrenstorfer公司);内标:13C6-克百威(100 mg/L,纯度≥99%,美国 Cambridge Isotope Lab,Inc.)。
混合标准溶液的配制:分别称取10.0 mg(精确到0.01 mg)氨基甲酸酯类农药标准品,置于10 mL棕色容量瓶中,用乙腈溶解并定容至刻度,配成质量浓度为1 000 mg/L的单标准储备溶液,于-20℃冰箱中保存。根据实际需要分别取适量的单标准储备液用乙腈稀释混合后配制成混合标准中间液。取适量混合标准中间液用初始流动相配制系列标准工作液。
同位素内标标准溶液的配制:准确移取1.0 mL13C6-克百威标准溶液(100 mg/L)于10 mL棕色容量瓶中,用乙腈定容至刻度,配成10 mg/L的内标储备液,于-20℃冰箱中保存。使用前用乙腈逐级稀释为1.0 mg/L的同位素内标工作溶液。
1.2 样品前处理
1.2.1 样品提取
称取试样2.0 g(精确至0.01g)于50 mL具塞聚丙烯离心管中,加入20 μL内标工作溶液涡旋振荡30 s混匀,加入 1 g NaCl、4 g MgSO4,混匀后加入20 mL乙腈(经正己烷饱和),涡旋振荡30 s混匀后,于20℃超声提取30 min,在6 000 r/min离心5 min;上清液(乙腈层)转移至另一个50 mL具塞聚丙烯离心管中,样品管中加入20 mL乙腈(经正己烷饱和)再次超声提取10 min,于6 000 r/min下离心5 min后,转移乙腈层,与第一次的上清液合并。提取液经装有5 g无水硫酸钠的玻璃漏斗,滤纸过滤至150 mL鸡心瓶中,用20 mL乙腈分两次淋洗无水硫酸钠并收集滤液。样品提取液在35℃水浴中经旋转浓缩至近干,余下的溶剂用弱氮气流缓慢吹干。用适量环己烷-乙酸乙酯(1∶1,v/v)溶解残渣,并清洗鸡心瓶后转移至10 mL刻度离心管中,用环己烷-乙酸乙酯(1∶1,v/v)定容至刻度,于6 000 r/min下离心10 min,上层清液经0.22 μm 有机相微孔滤膜过滤于10 mL凝胶渗透色谱(GPC)专用试管中,待GPC净化。
1.2.2 样品净化
采用GPC进行净化,GPC净化柱管尺寸为300 mm×20 mm,内装25 g Bio-Beads S-X3填料(美国Bio-Rad公司);紫外检测波长为254 nm;流动相为环己烷-乙酸乙酯(1∶1,v/v);流速为 4.7 mL/min;进样量为5 mL。从10 min开始收集,收集时间为10 min。将收集液于35℃水浴中浓缩至近干,余下溶剂用弱氮气流缓慢吹干后用200 μL初始流动相溶解,经0.22 μm有机滤膜过滤于LC专用进样瓶中,待HPLC-LIT-MS分析。
1.3 液相色谱-质谱条件
1.3.1 液相色谱条件
色谱柱:SHISEIDO Capcell PAK CR柱(100 mm ×2.1 mm,5 μm;SCX-C18(1∶4));柱温:35℃;流动相:A液为含有5 mmol/L甲酸铵和0.1%(v/v)甲酸的水溶液,B液为乙腈;梯度洗脱条件:0~15 min,10%B ~70%B;15~20 min,70%B ~90%B;20~24.5 min,90%B;24.5~25 min,90%B~10%B;25~30 min,10%B;样品池温度:4℃;进样量:10 μL;流速:0.2 mL/min。
1.3.2 质谱条件
电喷雾电离正离子源(ESI+);喷雾电压4 kV;毛细管电压72 V;棱镜电压36 V;毛细管温度300℃;鞘气流速7 mL/min;辅助气流速15 mL/min;扫描方式:三级质谱选择反应监测(SRM);保留时间及质谱参数见表1。
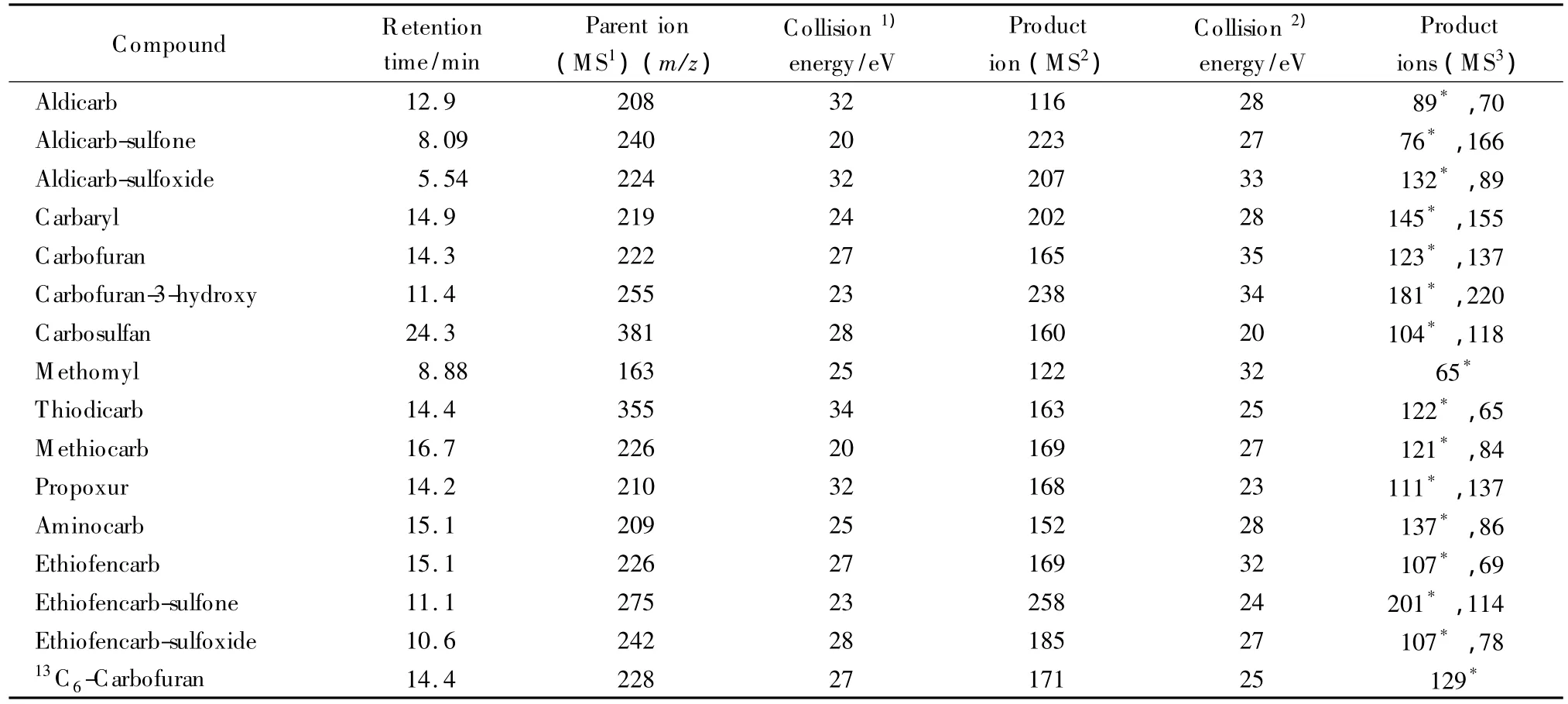
表1 氨基甲酸酯类农药保留时间和主要质谱参数Table 1 R etention times and main MS parameters of NMC pesticides
2 结果与讨论
2.1 液相色谱条件和质谱条件的优化
为考察在本实验条件下涕灭威、涕灭威砜、涕灭威亚砜、甲奈威、克百威、3-羟基克百威、丁硫克百威、灭多威、硫双灭多威、甲硫威、残杀威、灭害威、乙硫苯威、乙硫苯威砜、乙硫苯威亚砜及内标物的质谱行为,根据目标物的相对分子质量,在m/z 50~500范围内,分别用500 μg/L的单标准工作液进行流动注射分析,采用ESI源正离子扫描,先优化电喷雾电压、鞘气、辅助气,然后逐级优化各化合物的质谱参数,确定特征碎片离子及碰撞能量等质谱分析条件。结果见表1。
大多数氨基甲酸酯类农药的母体和降解产物在普通 C18柱上都有保留[9,12],但在本实验条件下多数氨基甲酸酯农药降解产物的色谱峰拖尾,峰形较差。SHISEIDO Capcell PAK CR是一款由反相填料C18和强阳离子(磺酸基团)填料混合填充而成的色谱柱,具有C18柱反相分离模式和磺酸基的强阳离子交换模式,因此在酸性条件下,有利于弱碱性化合物的保留及色谱峰形的改善。在已有工作[16]的基础上,我们考察了所有目标物在 SHISEIDO Capcell PAK CR 柱(100 mm ×2.1 mm,5 μm;SCX-C18(1∶4))上的色谱行为,结果表明,降解产物在CAPCELL PAK CR柱上的峰形优于在普通C18柱上的峰形,因此选用CAPCELL PAK CR色谱柱为分析柱。
2.2 提取和净化条件的选择
乙腈是提取氨基甲酸酯类农药的常用试剂,由于膳食样品和花生同属油脂含量高的样品,因此本实验参照已建立的花生样品的提取净化方法[16],用正己烷饱和后的乙腈作为提取溶剂,GPC净化去除样品中油脂和色素等大分子基质的干扰。GPC净化条件以油脂含量最高的肉类样品为研究对象,从9.5 min开始收集馏分,但发现馏分蒸干后仍有少量油脂。改为从10 min开始收集,馏分中基本没有油脂残留。因此确定所有样品基质都从10 min开始收集GPC馏分,收集时间为10 min。从回收率结果看,选定的GPC实验条件能够保证在有效净化样品的前提下收集大部分目标物。
2.3 基质效应
基质效应可以用基质效应因子(matrix factor,MF)来表示,MF=A/B×100%。A为目标物在纯溶剂中的响应值,B为在空白样品基质(蔬菜类)中添加相同水平目标物的响应值,实验结果见表2。
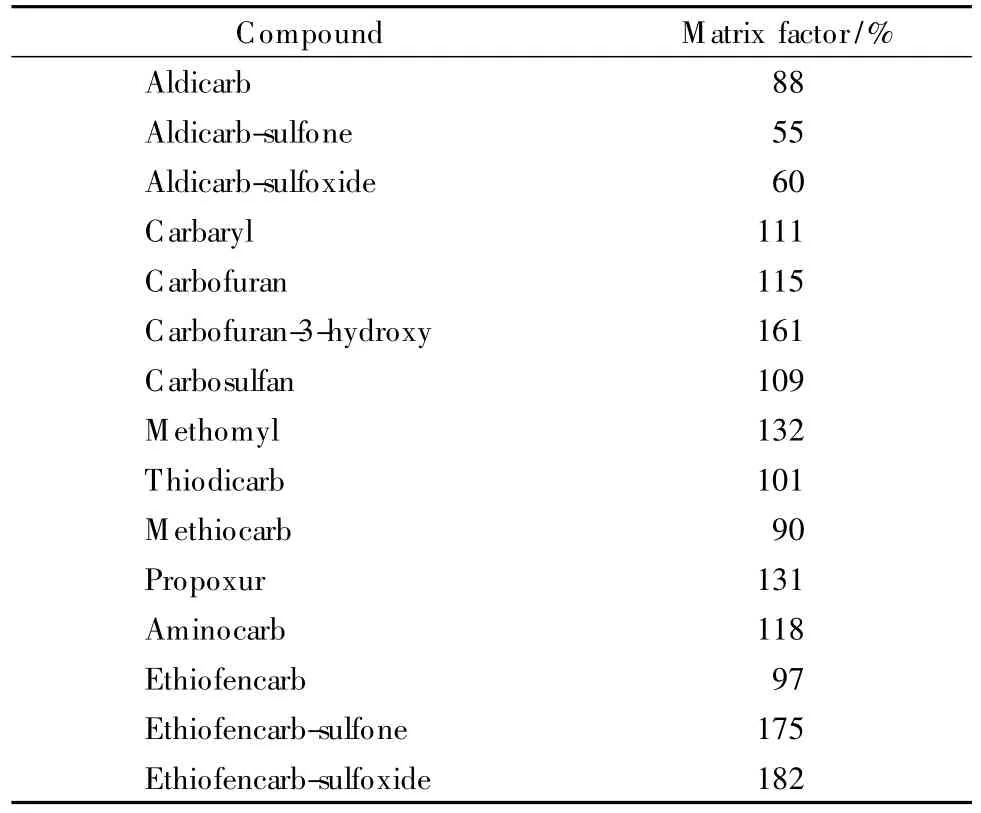
表2 氨基甲酸酯类农药在蔬菜类样品中的基质效应Table 2 Matrix effect of NMC pesticides in the vegetable samples
一般而言,MF>150%为基质抑制,MF <70%为基质增强。表2结果表明,基质效应对降解产物的影响比较大,其中对涕灭威砜、涕灭威亚砜有基质增强作用,对3-羟基克百威、乙硫苯威砜和乙硫苯威亚砜有基质抑制作用,对农药母体的影响相对较小。由于同一目标物在不同基质中所受的基质效应的大小不同,为降低基质效应对目标物的影响,应使用相应的空白基质配制标准曲线。
2.4 定性依据及定量方法
本文采纳欧盟2002/657/EC[17]规定的质谱定性确证要求,即采用低分辨质谱进行定性确证分析时需要4分(每个母离子1分,每个子离子1.5分,总共4分);进行实际样品测定时,如果样品质量色谱峰的保留时间与标准溶液的目标组分的保留时间一致(变化范围在±2.5%之内),而且质量色谱峰的离子相对丰度比与浓度接近的标准溶液的离子相对丰度比偏差在±20%之间,则可判定样品中存在目标化合物。每个目标物均以13C6-克百威为内标物,采用内标法定量。
2.5 标准曲线、回收率、精密度和检出限
在选定的实验条件下,采用空白蔬菜类和肉类样品进行加标回收试验,分别做低、中、高3个加标水平,每个浓度水平平行做5份样品,计算平均加标回收率及相对标准偏差(RSD)。各目标物平均加标回收率在60.4%~114%之间,相对标准偏差在3.46%~16.2%之间,结果见表3。以平均加标回收率表示方法的准确度,以平均加标回收率的RSD表示方法的精密度。尽管个别化合物(灭害威)或代谢产物(3-羟基克百威)的回收率偏低(60.4%),但从加标回收实验结果看,总体来说该方法的准确度及精密度符合多残留分析的要求。各目标物在空白蔬菜类样品基质中的质量色谱图见图1。
用混合标准工作液,以空白基质为稀释液配制标准系列溶液(内标浓度均为100 μg/L)。以各组分定量离子色谱峰面积与内标物色谱峰面积之比乘以内标的浓度为纵坐标,以各目标组分的浓度为横坐标绘制标准曲线,各组分线性关系良好,相关系数均大于0.991 6,结果见表4。
用蔬菜类样品低加标水平计算各组分的检出限(LOD)和定量限(LOQ):S/N=3对应的含量为LOD,S/N=10对应的含量为LOQ。各组分在蔬菜类样品中的检出限在0.001~0.010 mg/kg范围内。结果见表4。
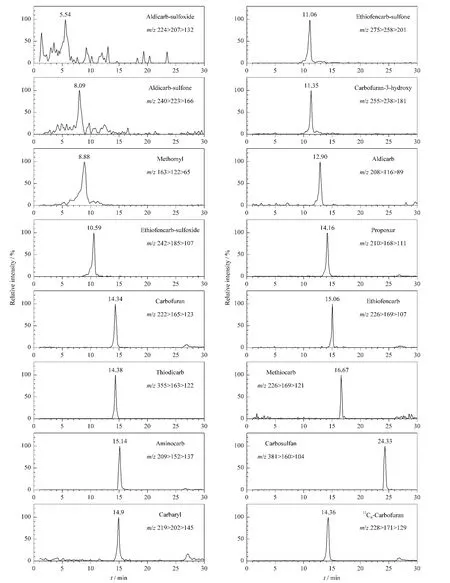
图1 空白蔬菜类样品中氨基甲酸酯类农药加标(20 μg/kg)的质量色谱图Fig.1 SR M chromatograms of NMC pesticides in blank vegetable samples(spiked level of 20 μg/kg)
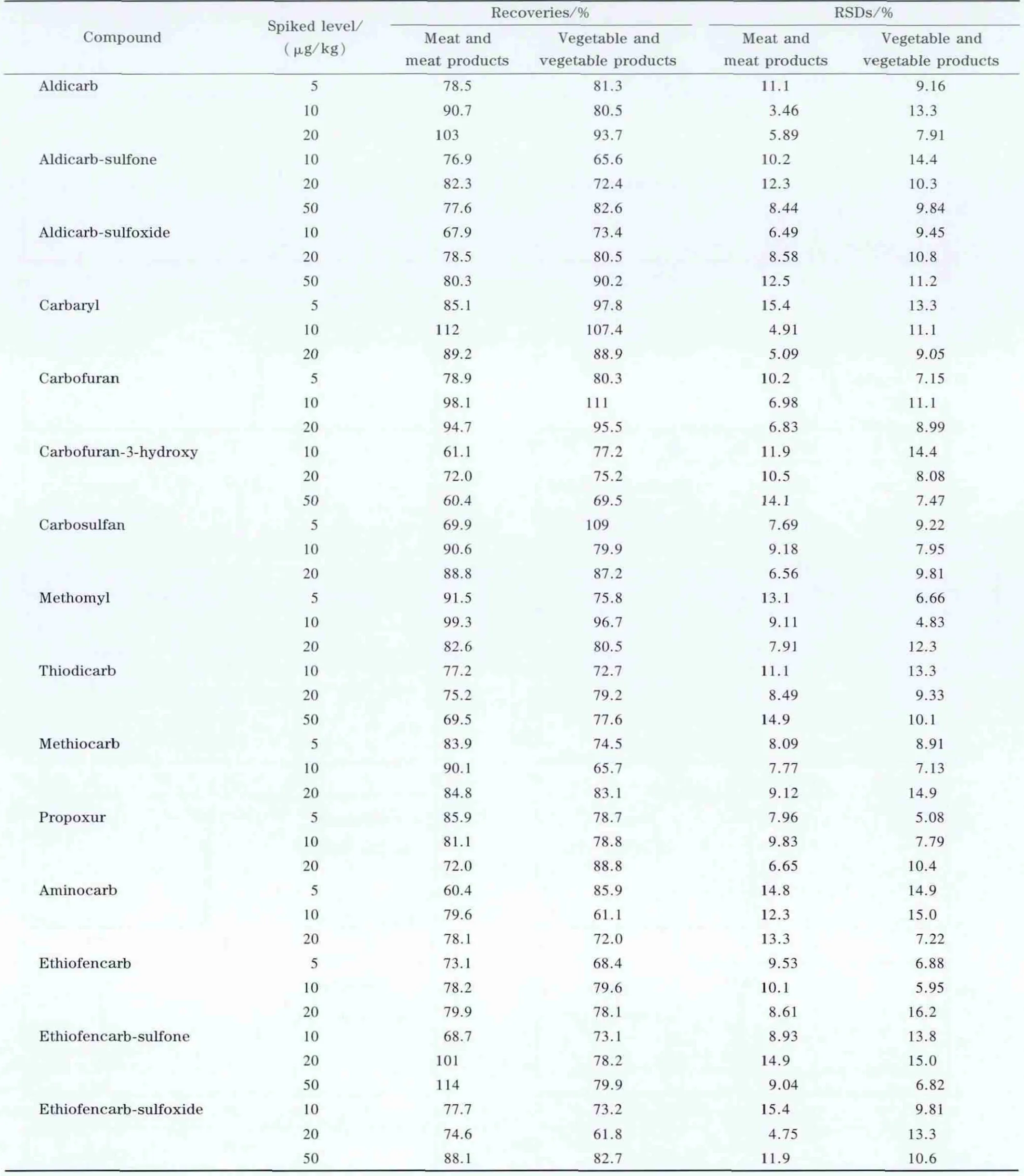
表3 蔬菜类和肉类样品中氨基甲酸酯类农药的加标回收率及精密度(n=5)Table 3 R ecoveries and R SDs of NMC pesticides in blank meat and meat products/vegetable and vegetable products(n=5)
2.6 实际样品测定
应用所建立的方法对2007年第四次中国总膳食研究项目的9类样品基质(肉类、蛋类、乳类、水果类、蔬菜类、谷类、豆类、薯类、水产类)共108份样品中的氨基甲酸酯类农药进行了检测,结果如下:检出了涕灭威和克百威,检出率分别为5.6%和8.3%,且其在蔬菜类样品中的含量较高,涕灭威的最高含量为105 μg/kg,克百威的最高含量为3.56 μg/kg(见图2);未检出本文所考察的其他氨基甲酸酯类农药。
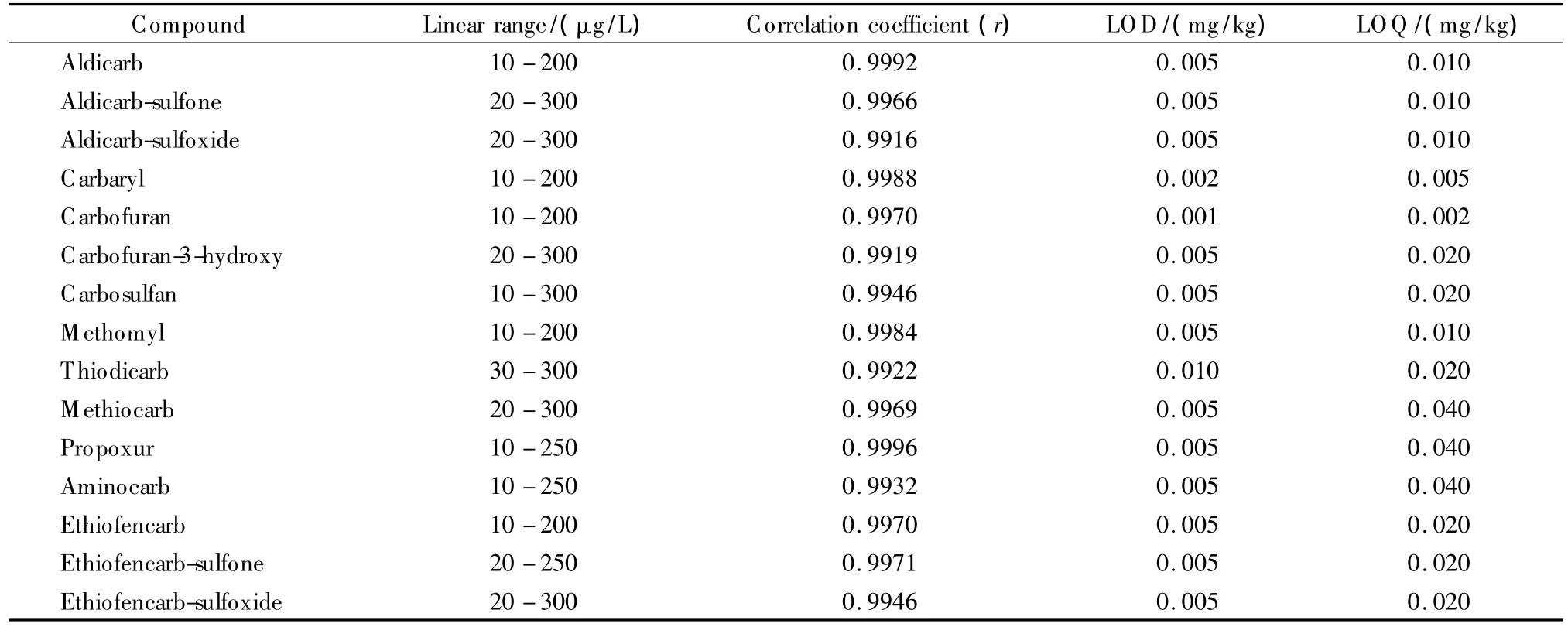
表4 氨基甲酸酯类农药的线性范围、相关系数、检出限及定量限Table 4 Linear ranges,correlation coefficients,LODs and LOQs of NMC pesticides
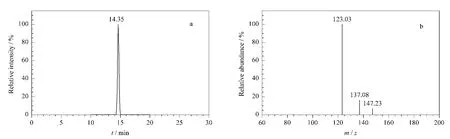
图2 蔬菜类样品中检出的克百威的(a)质量色谱图及(b)三级质谱图Fig.2 (a)MR M chromatogram and(b)MS3spectrum of carbofuran in a real vegetable sample
3 结论
本文以膳食样品为研究对象,建立了高效液相色谱-串联线性离子阱质谱法(HPLC-LIT-MS)测定氨基甲酸酯类农药的检测方法,为氨基甲酸酯类农药的残留监控和风险评估等研究提供了一种准确、可靠的分析手段。
[1] Karami-Mohajeri S,Abdollahi M.Hum Exp Toxicol,2011,30(9):1119
[2] World Health Organization(WHO).Acute Hazard Exposure Assessment for Chemicals in Food.[2013-12-31].http://www.who.int/foodsafety/chem/acute_data/en/print.html
[3] Jensen A F,Petersen A,Granby K.Food Addit Contam,2003,20(8):776
[4] Rawn D F K,Roscoe V,Krakalovich T,et al.Food Addit Contam,2004,21(6):555
[5] London L,de Grosbois S,Wesseling C,et al.Int J Occup Environ Health,2002,8(1):46
[6] Codex Alimentarius Commission.FAO/WHO Food Standard.[2013-12-31].http://www.codexalimentarius.net/pestres/data/pesticides/details.html?id=117
[7] Santos Delgado M J,Rubio Barroso S,Fernadez-Tostado G T,et al.J Chromatogr A,2001,921:287
[8] Wang X,Li P,Zhang W,et al.J Sep Sci,2012,35(13):1634
[9] Martinez Fernandez J,Parrile Vazquez P,Martinez Vidal J L.A-nal Chim Acta,2000,412:131
[10] Soriano J M,Jimenez B,Font G,et al.Crit Rev Anal Chem,2001,31(1):19
[11] Makihata N,Kawamoto T,Teranishi K.Anal Sci,2003,19:543
[12] Fernandez M,Pico Y,Manes J.J Chromatogr A,2000,871:43
[13] Sun J,Dong T,Zhang Y,et al.Anal Chim Acta,2010,666(1/2):76
[14] Wu C C,Chu C,Wang Y S,et al.J Environ Sci Health B,2008,44(1):58
[15] Li X W,Wu Y N,Chen J S.Chinese Journal of Epidemiol(李筱薇,吴永宁,陈君石.中华流行病学杂志),2011,32(5):456
[16] Yang X,Li P,Zhao Y F,et al.Chinese Journal of Chromatography(杨欣,李鹏,赵云峰,等.色谱),2012,30(3):314
[17] Commission Decision 2002/657/EC
