高效液相色谱-四极杆飞行时间质谱法同时检测豆芽中的3种外源植物激素残留
2014-05-08谢寒冰周明莹赵海峰王毅刚蒋万枫
谢寒冰, 周明莹, 赵海峰, 王毅刚, 蒋万枫, 赵 珊
(1.青岛市“菜篮子”商品质量监督检测中心,山东青岛266071;2.中国水产科学研究院黄海水产研究所,山东青岛266071)
豆芽是深受我国居民喜爱的一类食品,但近年来问题豆芽屡有报道,一些小作坊在生产过程中滥用添加剂,影响其质量安全。6-苄基腺嘌呤(6-BA)、4-氯苯氧乙酸(4-CPA)、赤霉素(GA3)等 3 种外源激素是问题豆芽培育过程中较常用的“AB粉”、“无根豆芽素”等的主要成分。4-CPA属内吸性植物生长调节剂,能调节植物株内激素的平衡,刺激子房膨大,补充植物体内生长素不足,促进生长[1],作为一种添加剂在培育无根豆芽时使用[2]。GA3和6-BA在种子萌发和休眠中的调控作用已得到广泛认可[3]。GA3可在植物幼苗生长过程中提高发芽率。6-BA与植物内源激素结构性质相似[4],在豆芽生长过程中可以促进芽的形成、抑制根的生长[3]。人体摄入过多6-BA会刺激皮肤黏膜,出现恶心、呕吐等现象[5]。国家质检总局159号公告明确指出6-BA、4-CPA不得作为添加剂生产和使用。我国尚未制定水果、蔬菜中赤霉素最高残留限量国家标准。针对这3种化合物的检测方法主要有高效液相色谱法[6-11]、高效液相色谱-质谱法[5,12,13]以及串联质谱法[14-16]、气相色谱法[17]、离子色谱法[18]和薄层色谱法[19],但这些方法只能检测3种目标化合物中的一种或两种。目前尚未见到国内运用高分辨质谱同时检测这3种外源激素的报道。
本文以黄豆芽和绿豆芽为样品,采取QuECh-ERS(quick,easy,cheap,effective,rugged,and safe)前处理方法,运用溶剂分步提取、分散固相净化等技术进行样品前处理,应用液相色谱-四极杆飞行时间质谱同时检测3种外源激素药物残留,并建立了质谱检索筛查库。方法具有快速高效、高灵敏度等特点,为市场豆芽质量安全监控提供了新的检测技术选择。
1 实验部分
1.1 仪器试剂
高效液相色谱-质谱联用仪:1290二元泵液相系统和G6530A Q-TOF高分辨质谱系统(安捷伦公司,USA)。Sigma 3-30ks离心机(希格玛公司,德国);N-EVAP11氮吹仪(Organomation公司,USA)。Milli-Q去离子水发生器(Millipore公司,USA)。甲酸、甲醇、乙腈(HPLC级,Honeywell Burdick&Jackson)。赤霉素、6-苄基腺嘌呤、4-氯苯氧乙酸标准品(德国 Dr.Ehrenstorfer公司)。C18、N-丙基乙二胺(PSA)吸附剂(博纳艾杰尔公司);硅胶、硅藻土、中性氧化铝(国药集团化学试剂公司);无水硫酸钠、乙酸、盐酸、无水乙醇等均为国产分析纯试剂。
1.2 标准溶液配制
分别准确称取GA3、6-BA、4-CPA标准品10.0 mg置于10 mL棕色容量瓶中,用甲醇溶解并定容,配制成1.0 g/L的标准储备液,转入棕色样品瓶中,同时配制1.0 mg/L的标准溶液,-20℃下保存。实验时用空白豆芽样品提取液配制标准工作溶液,现用现配。
1.3 样品预处理
准确称取2 g(精确至0.01 g)打碎混匀后的豆芽样品于聚四氟乙烯离心管中。加入2 g无水硫酸钠、0.5 g氯化钠,充分搅拌去除水分;加入20 mL含1%(v/v,下同)乙酸、50%乙醇、49%乙腈的溶液,振荡混匀5 min;以9 000 r/min离心5 min,收集上清液于50 mL聚四氟乙烯离心管中。残渣中加入10 mL盐酸溶液(1 mol/L)和10 mL乙腈,振荡提取5 min,以9 000 r/min离心,收集上层乙腈溶液。合并两次提取液,在提取液中加入1.0 g硅藻土、10 mL正己烷,振荡混匀5 min,以9 000 r/min离心5 min后弃去正己烷层。剩余溶液在50℃水浴条件下氮吹至近干,加入2.0 mL 50%甲醇水溶液,振荡混匀,过0.22 μm滤膜后上机测定。
1.4 色谱条件
色谱仪:Agilent 1290 Infinity LC。色谱柱:ZORBAX Eclipse Plus C18(100 mm ×3.0 mm,1.8 μm);柱温:30℃。进样体积:10 μL。流动相:0.1%甲酸水溶液(A)和甲醇(B)。梯度洗脱程序:0~1.0 min,80%A;1.0~8.5 min,80%A 降到50%A;8.5~8.6 min,50%A 降到1%A;8.6~13.0 min,保持1%A;13.0~13.1 min,1%A升到80%A;13.1~15.0 min,保持80%A。流速:0.4 mL/min。
1.5 质谱条件
质谱仪:Agilent 6530 Accurate-Mass Q-TOF;离子源:双喷射离子源(Dual AJS ESI);雾化气压强:275.8 kPa;干燥气温度325℃,流速8 L/min;鞘气温度350℃,流速11 L/min。负离子模式:毛细管电压3 000 V;毛细管出口电压(fragmentor)130 V;锥孔电压(skimmer)65 V;八极杆电压750 V。采集模式:targeted MS/MS。一级质谱扫描范围m/z 100~500,采集频率5 spectra/s;二级质谱扫描范围m/z 50~500,采集频率 10 spectra/s,窗口时间 0.4 min,碰撞池能量30 V;四极杆分辨率Medium m/z 4。参比离子m/z 112.985 5、301.998 1。质谱调谐:负离子模式自动调谐;质量范围:低于m/z 1 700;扩展动态范围2 GHz。
1.6 定性定量分析
targeted MS/MS模式下,同时对3种目标化合物进行一级、二级质谱全扫描,通过化学工作站软件建立3种目标化合物一级、二级质谱全扫描筛查库。定性分析时,根据目标化合物的一级、二级质谱精确质量数、保留时间、同位素丰度匹配、特征二级离子等检索确认。定量分析时,以准分子离子[M -H]-峰面积外标法计算,提取质量数宽度为15 ppm。
2 结果与讨论
2.1 色谱条件
为得到更好的色谱分离和质谱离子化效率,本文参考有关标准和文献方法[2,4,5,14-16],以甲醇-水作为流动相体系。水相中分别添加0.1%甲酸和5 mmol/L甲酸铵,用这两种流动相体系和Agilent Poroshell 120 EC-C18(100 mm ×3.0 mm,2.7 μm)、Agilent ZORBAX Eclipse Plus C18(100 mm×3.0 mm,1.8 μm)、Agilent Eclipse XDB-C18(150 mm ×2.1 mm,3.5 μm)3种色谱柱进行交叉实验。结果显示,流动相为甲醇-水(0.1%甲酸)、色谱柱为Agilent ZORBAX Eclipse Plus C18柱时,液相色谱流速设置可满足质谱检测要求,质谱离子化效率较高,3种目标物的色谱分离对称、峰形尖锐。图1为在以上优化条件下3种目标化合物的准分子离子[M -H]-提取色谱图。
2.2 质谱条件
2.2.1 检测模式与定量离子
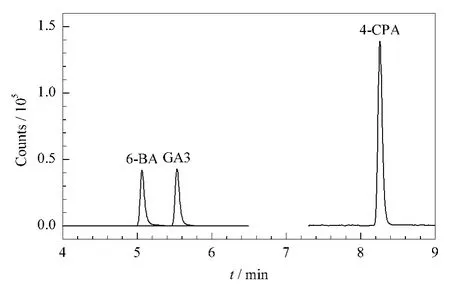
图1 3种目标化合物的提取色谱图Fig.1 Extracted ion chromatograms of the three target compounds
运用Agilent G6530A Q-TOF高分辨质谱分别对3种目标化合物进行一级质谱正、负离子模式全扫描,结果GA3和4-CPA在负离子模式下有强响应,6-BA在正、负离子模式下均有较好响应。综合考虑同时检测3种目标化合物的仪器参数设置要求、最佳信噪比等因素,采用负离子检测模式。负离子模式检测的基质干扰相对正离子模式要小,高分辨质谱精确质量测定也能进一步降低干扰,同时由于二级质谱灵敏度要小于一级质谱,因此本方法采用准分子离子峰面积定量。
2.2.2 诱导解离电压和碰撞电压
质谱仪的毛细管出口区域碰撞诱导解离电压影响离子传输效率,进而影响检测灵敏度。考察了其分别为70、100、130、160和200 V 时3种目标化合物的响应值(见图2)。GA3和6-BA的峰面积随电压升高而逐渐增大;4-CPA的峰面积在130 V前随电压升高而增大,在超过130 V后随电压升高而减小。为保证各目标物均能得到较强响应,确定毛细管出口区域碰撞诱导解离电压为130 V。
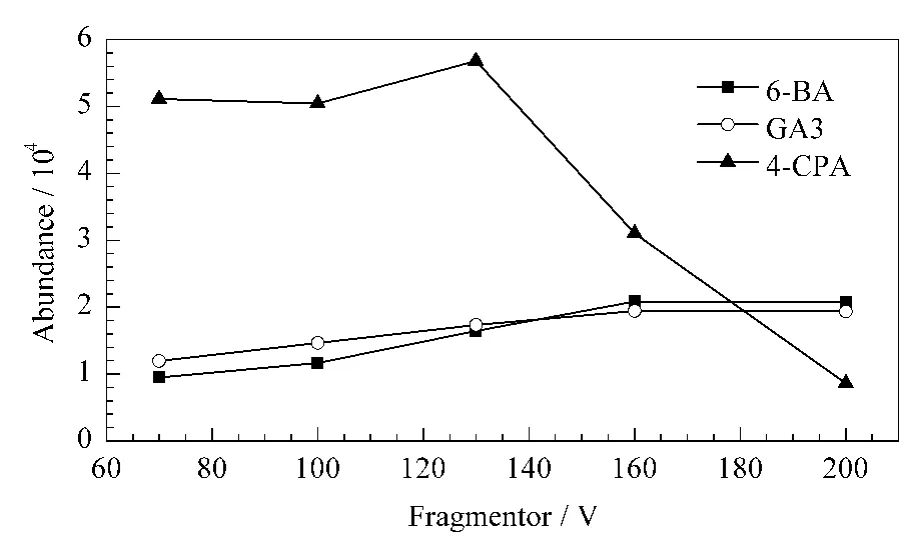
图2 不同碰撞诱导解离电压条件下目标物的质谱响应值Fig.2 R esponses of the target compounds under different fragmentor conditions
另外,碰撞池电压决定母离子碎裂程度[20],影响二级质谱响应值和特征峰匹配。考察了各目标物分别在10、20、30和40 V碰撞电压条件下的二级质谱。当碰撞池电压为30 V时,各目标物均能得到较好的二级质谱特征信息(见图3),因此选择30 V为本方法中碰撞池电压。
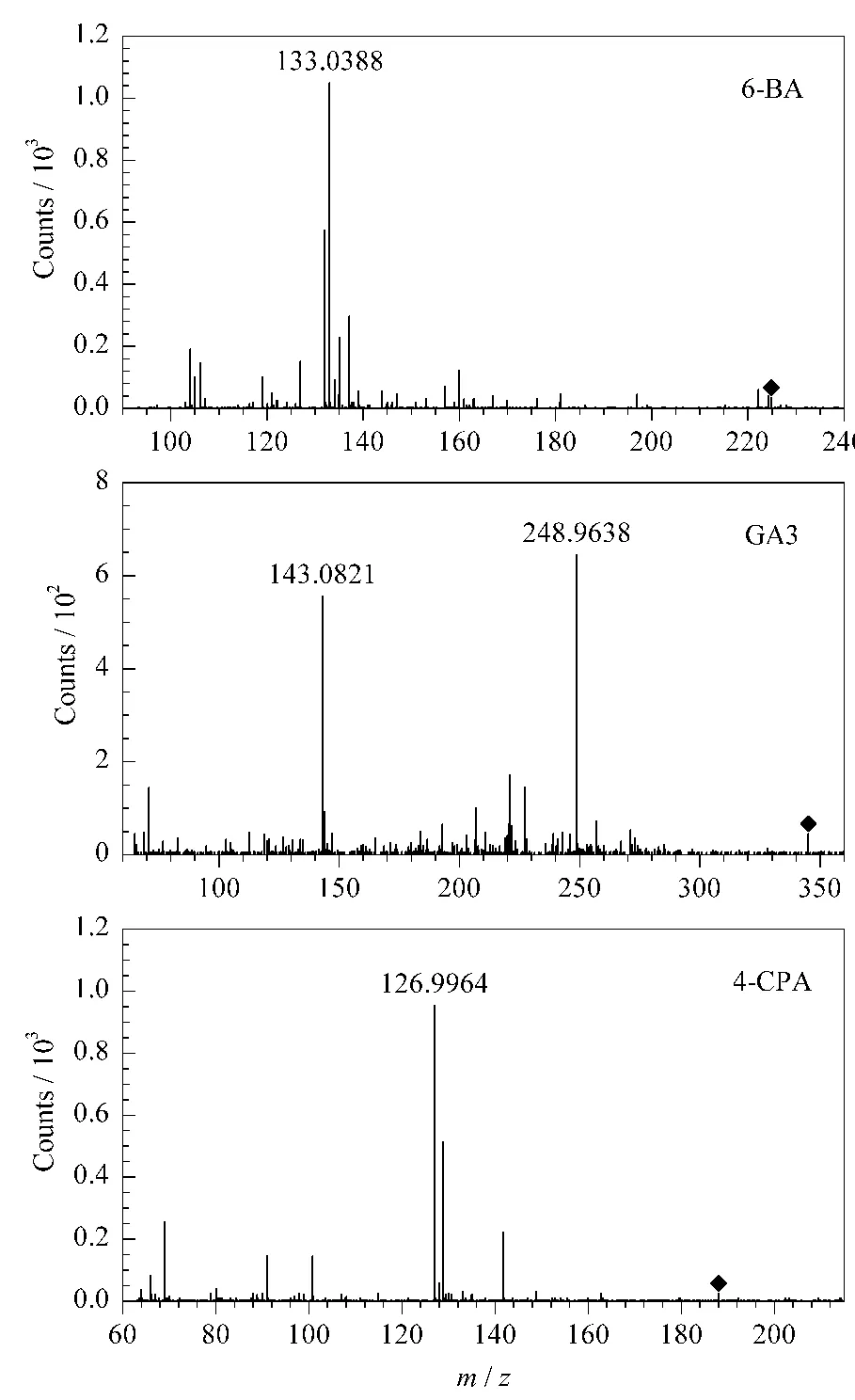
图3 碰撞池电压为30 V时3种目标化合物的二级质谱图Fig.3 MS/MS spectra of the three target compounds(collision energy:30 V)
2.2.3 二级质谱
targeted MS/MS模式可一次进样同时检测3种目标化合物的一级和二级质谱全扫描信息。在化学工作站设置3种目标化合物二级质谱检测的母离子及保留时间等信息,同时设置时间窗口、碰撞能量和参比溶液等信息,检测得到的二级质谱特征离子如表1所示。
2.3 提取净化
3种目标物具有不同的化学性质:6-BA难溶于水,可溶于酸、碱,在酸性、碱性条件下均稳定;4-氯苯氧乙酸在样品中多以4-氯苯氧乙酸钠形式存在,4-氯苯氧乙酸钠易溶于水、碱性水溶液,酸化后生成4-氯苯氧乙酸,易溶于乙醚、乙醇等有机溶剂;GA3难溶于水,能溶于醇、酮、酯类等有机溶剂。
据此,本方法采用QuEChERS前处理技术,对比3种不同方式的回收效果:酸化乙腈(含1%乙酸)一次性提取、酸化乙醇-乙腈(1%乙酸+50%乙醇+49%乙腈)一次性提取、酸化乙醇-乙腈(1%乙酸+50%乙醇+49%乙腈)提取后再在酸化条件下以乙腈盐析二次提取。结果酸化乙醇-乙腈溶液效果较好,但对4-CPA的回收效率不高,主要原因是仍有4-CPA以钠盐形式存在于试样残渣中,没有被有机溶剂提出。在以酸化乙醇-乙腈提取后的残渣中加入稀盐酸溶液和乙腈,利用盐析分层在酸性条件下以乙腈溶剂提取4-CPA,结果4-CPA回收效率得到有效改善。在以酸化乙醇-乙腈溶液提取目标物时,必须加入无水硫酸钠、氯化钠除去试样中的水分,因为水的存在会严重影响各目标物的回收效率,且无水硫酸钠、氯化钠也是下一步乙腈盐析分层提取的必要试剂。另外测试了乙醇-乙腈提取液中乙醇体积分数变化对回收效果的影响。当乙醇含量增加时会增多基质干扰物的提取,但其含量过低则不利于GA3的提取,乙醇体积分数在50%时能满足回收效果和基质净化需求。在50 μg/kg添加水平下,各目标物在3种不同提取条件时的添加回收率如图4所示,分两步提取时各化合物的添加回收效果最优。
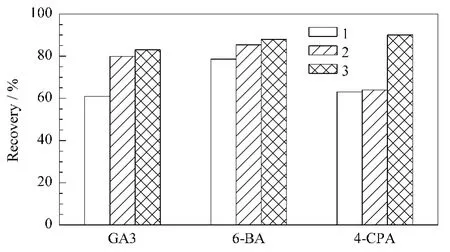
图4 不同提取液对豆芽中目标物的提取效果Fig.4 Effect of different extraction solutions on the target compounds in been sproutsExtraction solutions:1.1%acetic acid+99%acetonitrile;2.1%acetic acid+50%ethanol+49%acetonitrile;3.step one:1%acetic acid+50%ethanol+49%acetonitrile and step two:diluent hydrochloric acid+acetonitrile.
考察了C18、PSA、硅藻土、硅胶、氧化铝5种分散剂的净化效果。C18、PSA、硅胶、氧化铝等4种分散剂分别不同程度地存在吸附目标物或基质干扰物去除效果不好等问题;而硅藻土没有吸附目标化合物现象,对样品色素等基质干扰物的净化效果也优于其他试剂。硅藻土用量为0.2~1.5 g时,均未发生目标化合物吸附现象。从实际净化效果看,硅藻土吸附剂可净化样品提取液中的部分色素类物质,当其用量高于1.0 g后,增加分散剂用量对基质净化效果无明显改善。因此确定硅藻土用量为1.0 g。
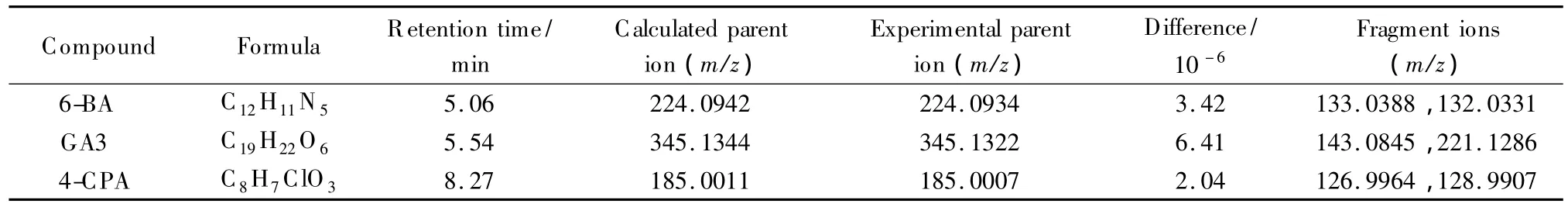
表1 3种目标物的保留时间和质谱信息Table 1 R etention times and MS information of the three target compounds
2.4 基质效应
基质物质在色谱分离过程中与目标化合物共流出,可能影响目标化合物的离子化过程,进而影响定量、线性、精密度等[21]。豆芽样品经过前处理过程后,提取液在提取目标化合物的同时也会提出豆芽内源基质物质和前处理过程中引入的外源基质物质。本方法考察了豆芽基质对3种目标化合物的质谱影响,用空白豆芽提取液配制10 μg/kg的目标化合物标准液,用甲醇-水(5∶5,v/v)溶液配制同等浓度的标准溶液,计算两者质谱响应强度的比值。结果显示 6-BA、Ga3、4-CPA的比值分别为 0.97、1.08、0.68。虽然固相分散剂的使用在一定程度上降低了基质抑制,但仍不能彻底解决。为此,本方法采用基质提取液配制标准工作溶液进行校正补偿[22],以进一步降低基质抑制对检测结果的干扰。
2.5 方法学考察
2.5.1 线性关系
取3种目标物标准溶液,以空白基质溶液配制成6种质量浓度的系列梯度(1~200 μg/L),按照1.4节和1.5节条件检测。以准分子离子峰[MH]-的色谱峰面积(y)作为纵坐标,以相应质量浓度(x,μg/L)作为横坐标,计算得到各目标物的线性方程,其相关系数以及线性范围等如表2所示。

表2 3种目标物的线性范围、回归方程和相关系数(r)Table 2 Linear ranges,regression equations and correlation coefficients(r)of the three target compounds
2.5.2 回收率、稳定性和定量限
以黄豆芽和绿豆芽为检测试样,按照本方法进行3个水平的添加回收试验,以6次平行测定结果计算回收率、相对标准偏差等,并按信噪比(S/N)为10确定定量限,结果见表3。3种目标化合物的回收率为79.1%~96.1%,相对标准偏差为5.7%~10.4%。GA3和 6-BA的定量限(LOQ)为 5.0 μg/kg,4-CPA 的 LOQ 为10.0 μg/kg。

表3 3种目标化合物的回收率、定量限和精密度(n=6)Table 3 R ecoveries,limits of quantification(LOQs)and relative standard deviations(R SDs)of the three target compounds(n=6)
2.6 方法的应用
应用本方法对从市场抽取的15批次黄豆芽、绿豆芽样品进行检测,检出1批次绿豆芽中6-BA含量为0.24 mg/kg,1批次黄豆芽中 GA3含量为0.19 mg/kg,1批次黄豆芽中4-CPA含量为0.49 mg/kg。分别应用 DB33/T 625.3-2007和 SN/T 0 350-2012方法对检出6-BA、GA3的两份样品进行测定,测得 6-BA、GA3含量分别为 0.28 mg/kg、0.16 mg/kg,与本方法结果一致。用不含3种目标化合物的黄豆、绿豆培育10批豆芽样品,应用本方法对其进行检测分析,均未检测出3种目标化合物。结果证明本方法的有效性。
3 结论
本文建立了应用QuEChERS快速前处理技术和液相色谱-飞行时间质谱检测豆芽中3种外源植物激素的方法,具有快速高效、定性准确、灵敏度高等特点,能够满足国内外关于豆芽中非法添加物残留限量的要求。本方法前处理技术在目标物提取效率、基质净化效果、方法稳定性等方面具有一定优势;采用的高分辨质谱检测技术可进行一级、二级质谱精确质量数全扫描,在目标化合物定性确认方面具有显著优势;在定量方面,本方法具有较好的稳定性,定量限能够满足实际工作需要,为市场豆芽质量安全筛查提供了新的检测方法选择。
[1] Geng G C,Pan Q M,Qian G W.Jiangsu Chemical Industry(耿国彩,潘启民,钱光伟.江苏化工),1995,23(4):47
[2] Feng J W,Wu J S,Cao G Y,et al.Physical and Chemical Testing Part B:Chemical Analysis(冯家望,吴洁珊,曹桂云,等.理化检验:化学分册),2012,48(10):1219
[3] Ding J Z,Yin T,Yu X,et al.Plant Physiology Journal(丁俊胄,尹涛,余翔,等.植物生理学报),2011,47(5):501
[4] Xie J,Zhang Y Z.Journal of Instrumental Analysis(谢君,张义正.分析测试学报),2001,20(1):60
[5] Jin M C,Chen X H,Li X P,et al.Analytical Instrumentation(金米聪,陈晓红,李小平,等.分析仪器),2005(3):29
[6] DB11/T 379-2006
[7] DBS22/001-2013
[8] GB/T 23381-2009
[9] Lin L.The Food Industry(林琳.食品工业),2008(3):65
[10] Huang W P.China Preventive Medicine(黄卫平.中华预防医学杂志),2002,36(1):44
[11] Peng S,Zhang H,Huan Z Y,et al,Shandong Agricultural Sciences(彭姝,张昊,郇正玉,等.山东农业科学),2010(2):95
[12] Li X P,Chen X H,Yao X P,et al.Chinese Journal of Health Laboratory Technology(李小平,陈晓红,姚浔平,等.中国卫生检验),2005,15(2):149
[13] Xie H B,Lin H,Jiang W F,et al.Shandong Agricultural Sciences(谢寒冰,林洪,蒋万枫,等.山东农业科学),2008(2):92
[14] Liu H,Wu B,Yin Y,et al.Chinese Journal of Chromatography(柳菡,吴斌,殷耀,等.色谱),2013,31(1):22
[15] DB33/T 625.3-2007
[16] SN/T 0350-2012
[17] Kuang H,Hou Y X,Chu X G,et al.Chinese Journal of Analytical Chemistry(匡华,侯玉霞,储晓刚,等.分析化学),2006,34(12):1733
[18] Yan J L,Yan Y Q,Wang L,et al.Chinese Journal of Health Laboratory Technology(颜金良,颜勇卿,王立,等.中国卫生检验杂志),2006,16(10):1027
[19] Ding Y C,Jiang A X,Zhong M W.Chinese Journal of Public Health(丁友昌,姜爱香,钟鸣文.中国公共卫生),1997,13(7):77
[20] Lan F,Zhang Y,Yue Z F,et al.Journal of Instrumental Analysis(蓝芳,张毅,岳振峰,等.分析测试学报),2012,31(12):1471
[21] Xiang P,Shen M,Zhuo X Y.Journal of Instrumental Analysis(向平,沈敏,卓先义.分析测试学报),2009,28(6):753
[22] Wang L Q,He L M,Zeng Z L,et al.Journal of Chinese Mass Spectrometry Society(王立琦,贺利民,曾振灵,等.质谱学报),2011,32(6):321
