Molecular Dynamics Simulation for the Binary Mixtures of High Pressure Carbon Dioxide and Ionic Liquids*
2014-03-25徐君臣王松喻文徐琴琴
(徐君臣)(王松)(喻文)(徐琴琴)
WANG Weibin(王伟彬)and YIN Jianzhong(银建中)**
State Key Laboratory of Fine Chemicals, School of Chemical Machinery, Dalian University of Technology, Dalian 116024, China
Molecular Dynamics Simulation for the Binary Mixtures of High Pressure Carbon Dioxide and Ionic Liquids*
XU Junchen(徐君臣), WANG Song(王松), YU Wen(喻文), XU Qinqin(徐琴琴),
WANG Weibin(王伟彬)and YIN Jianzhong(银建中)**
State Key Laboratory of Fine Chemicals, School of Chemical Machinery, Dalian University of Technology, Dalian 116024, China
Molecular dynamics simulation with an all-atom force field has been carried out on the two binary systems of [bmim][PF6]-CO2and [bmim][NO3]-CO2to study the transport properties, volume expansion and microstructures. It was found that addition of CO2in the liquid phase can greatly decrease the viscosity of ionic liquids (ILs) and increase their diffusion coefficient obviously. Furthermore, the volume expansion of ionic liquids was found to increase with the increase of the mole fraction of CO2in the liquid phase but less than 35% for the two simulated systems, which had a significant difference with CO2expanded organic solvents. The main reason was that there were some void spaces inter and intra the molecules of ionic liquids. Finally, site to site radial distribution functions and corresponding number integrals were investigated and it was found that the change of microstructures of ILs by addition CO2had a great influence on the properties of ILs.
molecular dynamics simulation, carbon dioxide, ionic liquids, diffusion, microstructure
1 INTRODUCTION
Supercritical carbon dioxide (scCO2), room temperature ionic liquids (RTILs), CO2-expanded liquids (CXLs) and water are commonly recognized as “green solvents” [1-3]. ILs have high solubility for a wide range of inorganic, organic, and polymeric molecules and have the advantage of near-zero vapor pressure at ambient conditions [4-6]. Therefore, they can be considered as environmental benign solvents. Furthermore, the structure of ions can be changed to design and tune ILs for particular applications, which has greatly widened the applications of ILs [7, 8]. However, ILs have some disadvantages including high viscosity and poor diffusivity etc. Recently, ILs coupling with CO2has been used to realize the integrated operation of reaction-separation, which arouses great attention and research interest [9, 10]. The addition of CO2can improve the transport properties of ILs by reducing its viscosity and improving its diffusion coefficient while maintaining its chemical stability and excellent solvent properties [11-13]. CO2is nontoxic, nonflammable, inexpensive, and has moderate critical temperature and pressure (Tc=304.2 K, Pc=7.38 MPa). Moreover, CO2is highly soluble in ILs, while ILs are almost insoluble in CO2. Thus, the mixture solvents of IL-CO2can avoid cross-contamination of solvents and simplify the process of chemical reaction and separation. It is known that the physicochemical properties of the solvents are fundamental to chemical industry applications and these properties are usually obtained from experiments or simulations. However, there are several disadvantages of experimental method due to the high pressure and high cost. Equation of state (EOS) and empirical formulas can provide some data, but there are still many limitations. Under these circumstances, the research on these multi-phase systems by molecular dynamics (MD) simulation has become a hot topic in recent years [14-17].
Many experiments [18-21] have reported the phase behavior, thermodynamics properties and dissolution characteristics of IL-CO2systems. But they only focused on macroscopic description of the system properties and it was difficult to obtain the microscopic structural information. Some researchers have proposed the strange phenomena that the viscosity of ILs significantly decreases with the addition of CO2and the volumes of the mixtures increase slightly even though a large amount of CO2is dissolved in the liquid phase, but it is difficult to give the explanations by experimental method. At present, there are some literatures reported the properties of IL-CO2systems from the microscopic view of molecular interactions and molecular structures. Shim et al. [22] have reported equilibrium and non-equilibrium solvation structure and dynamics in the mixture of [BMI][PF6] and CO2via MD simulation. Balasubramanian et al. [23, 24] have investigated the microscopic structural and thermodynamics properties of [bmim][PF6] and [bmim][PF6]-CO2mixtures. Berne et al. [25] have studied the dissolution and diffusion properties of [bmim][PF6]-CO2systems and proposed the free space hypothesis to explain the low volume expansion of [bmim][PF6]-CO2systems with high mole fraction of CO2. Maginn et al. [26] have proposed that the solubility of CO2in ILs mainly depends on the type of anion. These reports have greatly developed the research of IL-CO2systems from microscopic view. However,most of these researchers have studied only a single IL-CO2system by MD simulation, and little attention has been paid to the influence of different types of IL on the properties of IL-CO2systems. Furthermore, it is not clear about the underlying reason of some unique properties of IL-CO2systems, such as the diffusion coefficient of ILs increases with the increase of mole fraction of CO2, the viscosity of ILs decreases with the increase of mole fraction of CO2and the volume expansion of ILs does not change significantly upon dissolution of large amounts of CO2. In this work, it is attempted to explain these unique properties from the microstructure changes of ILs by addition of CO2.
In order to investigate the different IL-CO2systems from the aspect of molecular interactions and molecular structures, the commonest used [bmim][NO3] and [bmim][PF6] were selected as model compounds, which have different types of anion and the same type of cation. Molecular dynamics simulation has been carried out on the two binary systems of [bmim][PF6]-CO2and [bmim][NO3]-CO2at different concentrations of CO2. The diffusion coefficient, viscosity, molar volume and volume expansion, the influence of the types of anion on the hydrogen bonds interactions and microstructures of the IL-CO2systems have been investigated.
2 SIMULATION METHODS
Successful molecular simulation mainly depends on the validity of the force field. Many force field models of ILs based on all-atom have been developed [27-29]. In this work, imidazolium cation force field parameters of [bmim] were taken from Wang’s force field [28], which was an all-atom force field optimized on the base of assisted model building with energy refinement (AMBER) [30]. Anions force field parameters of [PF6] and [NO3] were taken from the models developed by Wang [28] and Voth [31], respectively. An improved elementary physical models (EPM2 potential model) [32] was taken to describe CO2molecules, which has been successfully used to reproduce the pressure-volume-temperature (PVT) behavior and diffusion coefficient of scCO2fluid [33, 34].
In this work, bond interaction terms include the bond stretching potential Ub, valence angle bending potential Uθ, and dihedral angle torsion potential Uφ. The expression of potential energy items is as follows:
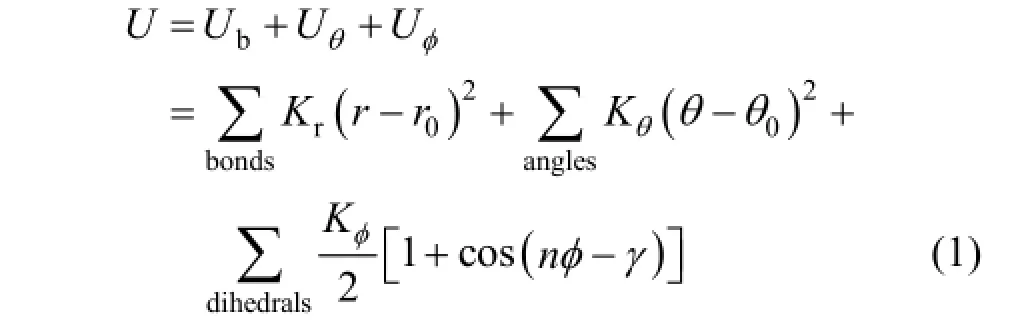
where Kris the bond force constant, which is a measure of the rigidity of the bond, r0is the equilibrium bond length, Kθis the angle bending force constant, θ0is the equilibrium angle, Kϕis the dihedral force constant, n is the periodicity, ϕ is the dihedral angle defined as the angle between the normal to the two planes, each of which is formed by three successive interaction sites, and γ is the equilibrium dihedral angle.
Nonbond interaction items (Unb) include van der Waals energy Uijand electrostatic energy Uelec. These interactions were calculated between only the atoms in different molecules or for the atoms in the same molecule separated by at least three bonds. Lennard-Jones 12-6 function was used as the expression of the van der Waals energy. The expression of nonbond interaction potential energy is as follows:

where εijis the traditional well-depth, σijand σmin,ijare the distances between atoms i and j, at which the energy of the two atoms reaches zero and minimum, respectively. Obviously,C=5.416069425×1030kJ·m·mol−1·C−2is a unit conversion factor and ε=1 is the relative dielectric constant. The qiand qjare the charges of atoms i and j, respectively, and rijis the distance between atoms i and j. The Lorentz-Berthelot mixing rules were employed to obtain the Lennard-Jones parameters for cross-interactions.

All systems were simulated at different CO2concentrations at 313.15 K. In comparison with the experimental data, CO2concentrations of [bmim][PF6]-CO2and [bmim][NO3]-CO2systems were set according to Han’s [35] and Brennecke’s [36] reports as shown in Table 1. In order to promote efficient equilibration, 100 ion pairs of ILs molecules were set in each system. For each system, all the corresponding molecules were added in a cubic box randomly with the initial density of 0.214 g·ml−1. All MD simulations were performed using the periodic boundary conditions.
First, each system was relaxed in the grand canonical ensemble (NVT ensemble) for 40 ps to remove the possible overlapping in the initial configuration and the temperature was set to 500 K. Second, to enable the volume variation, the equilibration run was performed in the isothermal-isobaric ensemble (NPT ensemble) for at least 1 ns, making sure that the system configuration was stable. Finally, the microcanonical ensemble (NVE ensemble) run was carried out for 5 ns to obtain the trajectory files in which the coordinates were recorded every 1 ps for further analysis. The main purpose of the relatively long simulation time is to ensure the systems to reach equilibrium andacquire the credible data. The pressure and temperature were maintained in the NPT ensemble using the Berendsen’s method [37] with the bath coupling time of 1 ps for both the thermostat and the barostat. The MD equations of motion were integrated using the Leap-frog algorithm with 2 fs time step. The calculation of the long-range interaction of coulomb forces was performed using the smooth particle Ewald (SPME) method [38]. The real space part of the Ewald sum and Lennard-Jones interactions were cut off at 1.5 nm. A 20934 kJ·mol−1energy deviation was accepted for all of the simulations.

Table 1 Parameter sets in this work
3 RESULTS AND DISCUSSION
3.1 Viscosity and diffusion coefficient
The chemical process using ILs as solvents is usually restricted by their high viscosity. The key problem for the applications of ILs in industry is to improve the transport properties by reducing its viscosity and improving its diffusion coefficient while maintaining its chemical stability and excellent solvent properties. The compressed CO2is an attractive reagent for reducing the viscosity of ILs [39] and it is beneficial to the applications in industry because CO2is environmental benign and can be easily separated from ILs by depressurization. The diffusion coefficient of MD simulation can be obtained by fitting the curves of the mean square displacement (MSD) vs. time. Usually, due to the strong hydrogen bonds interaction, the diffusion coefficient of the ILs is very low [39]. Many experimental reports [11, 21, 40] have investigated the diffusion coefficient of IL-CO2systems at low pressure (0.01-2.00 MPa). However, the viscosity of the systems decrease a little by addition of CO2at the low pressure and it is not beneficial to the industry applications. There are only a few experimental reports on the systems of IL-CO2at high pressure. For instance, Han et al. [35] have studied the viscosity of IL-CO2systems at the pressure ranged from 0.10 to 12.93 MPa and Brennecke et al. [36] have reported the phase behavior of IL-CO2systems at high-pressure (up to 9.57 MPa). They have made a contribution to the development of the research on coupling CO2with ILs, however, neither of them has reported the diffusion coefficient of IL-CO2at high pressure. Though there are many simulation reports [23-25] on the systems of IL-CO2at high pressure, relatively a few ones studied the transport properties of these systems using MD simulation. For the system of [bmim][PF6]-CO2, the diffusivity data of [bmim][PF6] reported by different scholars are not well confirmed each other [24, 25]. Therefore, the diffusion coefficient of the two systems was calculated by the Einstein relation [41].

where D is diffusion coefficient, Δr(t)2is the MSD of the center-of-mass of a molecule and “” represents ensemble average.
The long time MSD exhibits a linear behavior with respect to the time t. Herein, the diffusion coefficient D is obtained by a linear fitting of the slope in the region from 2 ns to 4 ns of MSD curves vs. time for all simulated systems. The results are given in Table 2. It can be seen that the diffusion coefficients of both [bmim][PF6] and [bmim][NO3] increase with the increase of the mole fraction of CO2in the liquid phase, which agrees with previous simulation results [24, 25]. When CO2mole fraction is 0.639 for [bmim][PF6]-CO2system, the diffusion coefficient of [bmim][PF6] in our simulation is close to Huang’s result and has great difference with Bhargava’s data (obtained by using the fitting cubic equation). This difference may be attributed to differences in the force fields and simulation time. When CO2mole fraction is 0.512, the diffusion coefficient of [bmim][NO3] is 61.40×10−12m2·s−1for [bmim][NO3]-CO2system and it is 4.5 times as highas that of the pure [bmim][NO3], while the diffusion coefficient of [bmim][PF6] is 59.30×10−12m2·s−1for [bmim][PF6]-CO2system with CO2mole fraction of 0.639 and it is 7.9 times as high as that of the pure [bmim][PF6]. Meanwhile, the diffusion coefficient of [bmim][NO3] has a significantly improvement with CO2mole fraction varying from 0.451 to 0.512. One reasonable explanation for this phenomenon is that the void spaces inter and intra IL molecules may be filled with CO2when CO2mole fraction is up to about 0.451. The structure of IL molecules has to significantly change in order to accommodate more CO2molecules, which has an obviously impact on the transport properties of ILs.
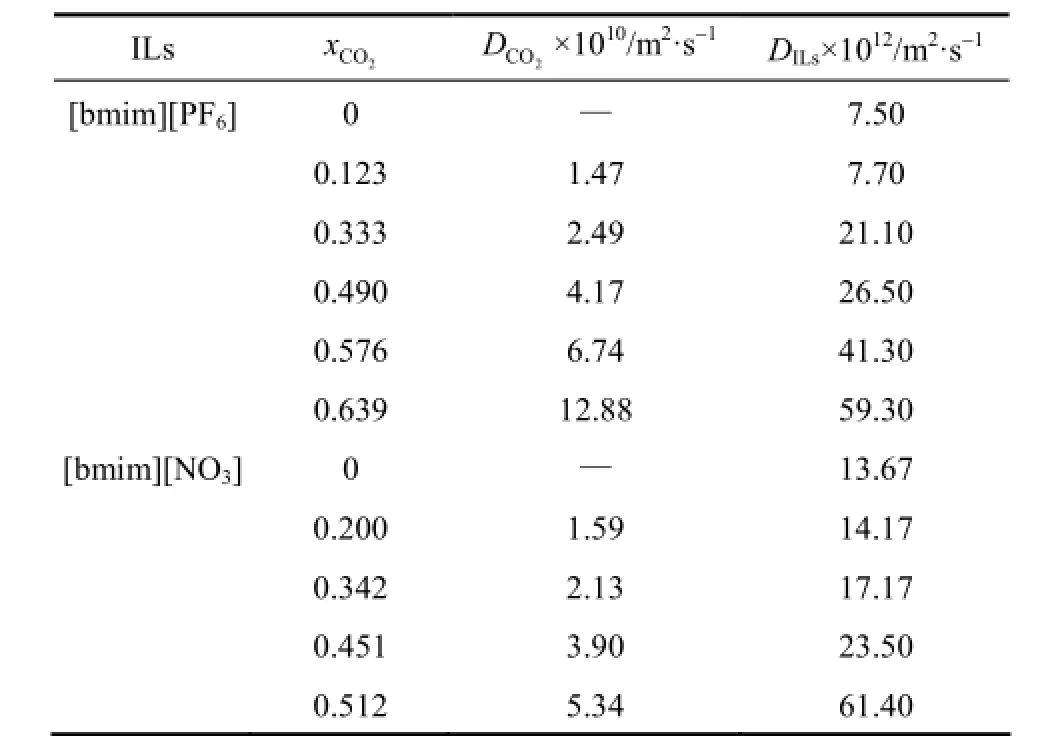
Table 2 Diffusion coefficient of ILs and CO2
The viscosity is calculated by the modified Stokes-Einstein relation [42] as shown in Fig. 1. It can be seen that the viscosity of both [bmim][NO3] and [bmim][PF6] decreases with the increase of the mole fraction of CO2and the simulation results of [bmim][PF6] are in good agreement with the experimental data [35] (no experimental data about [bmim][NO3]-CO2system). The viscosity of the pure [bmim][NO3] is 0.0633 Pa·s at 313.15 K, while it reduces to 0.0207 Pa·s when CO2mole fraction is 0.512 (the pressure of the system is 9.20 MPa). The viscosity of the pure [bmim][PF6] is 0.0923 Pa·s, while it reduces to 0.0172 Pa·s when CO2mole fraction is 0.639 (the pressure of the system is 10.93 MPa). Therefore, addition of CO2can significantly decrease the viscosity of ILs.

Figure 1 Viscosity of [bmim][NO3] and [bmim][PF6] with the increase of the mole fraction of CO2
Decrease of the viscosity is a focus of research on ILs and this will be helpful for its applications in some fields which are restricted by the high viscosity before. For example, direct immersion is a simple method of preparing supported ionic liquid membranes (SILMs), but it is time consuming and sometimes difficult to ensure whether ILs is effectively impregnated into the pores of supports due to its relatively high viscosity. For this reason, some researchers used ethanol or methanol to dilute the ILs to prepare SILMs [43, 44] because they can easily dissolve in ILs and greatly decrease its viscosity. However, organic solvents will cause secondary pollution and the membrane performance can be affected when ethanol or methanol is evaporated from ionic liquids. It is suggested that CO2should be applied to dilute the ILs and reduce the viscosity to prepare SILMs because CO2is benign to the environment and easy to separate from the products.
3.2 Volume expansion and molar volume
CO2is a well-known anti-solvent in gas anti-solvent process [45, 46] because the addition of it will lead to a significant increase in the volume of organic solvents. For example, a volume expansion greater than 1100% was obtained when the mole fraction of CO2was from 0 to 0.92 in acetonitrile [47]. Compared with organic solvents, ILs show slightly expansion in volume with CO2dissolution. Gallagher et al. [48] proposed a classical definition of volume expansion ratio ΔVr[Eq. (5)] based on the change in the absolute volume of the liquid.

where VLis total volume of the liquid mixture at a given temperature and pressure and V2is the volume of the pure liquid at the same temperature and ambient pressure. The volume expansion of two binary systems of [bmim][PF6]-CO2and [bmim][NO3]-CO2is calculated by Eq. (5), and the results are shown in Fig. 2.
As it can be seen in Fig. 2, the volume expansions of the two binary systems increase with the increase of the mole fraction of CO2. This trend is in good agreement with the experimental results presented by Brennecke et al [49]. Furthermore, the volume expansions of [bmim][PF6]-CO2and [bmim][NO3]-CO2are only 21.75% and 32.63% when CO2mole fraction are 0.512 and 0.639, respectively, which is much lower than that of CO2expanded organic solvents (i.e., for ethanol-CO2mixtures, the volume expansion is 90.40% with CO2mole fraction of 0.512) [50]. Usually, the increase of liquid volume is accompanied by a decrease in mixtures solvent strength. Brennecke et al. [19] reported the solvent strength of IL-CO2systems according to Kamlet-Taft parameters and found that the addition of CO2decreased the solvent strength of ILs to a very small extent. Thus, unlike in organic solvents, ILs do not expand to a great degree whenCO2is added, allowing ILs to maintain their solvent strength with the dissolution of large amounts of CO2. What deserved to be mentioned is that the volume expansion of the two mixtures of [bmim][NO3] and [bmim][PF6] has very little difference at the same CO2concentration. Brennecke et al. [49] measured volume expansion for sorts of IL-CO2systems and found it seems independent of the types of ILs although the molecular mass of these ILs varied a lot.

Figure 2 Volume expansion of [bmim][NO3] and [bmim][PF6] with the increase of the mole fraction of CO2
Liquid molar volume provides an insight into the unique phase behavior of IL-CO2systems. The molar volumes of two binary systems of [bmim][PF6]-CO2(CO2mole fraction from 0 to 0.639) and [bmim][NO3]-CO2(CO2mole fraction from 0 to 0.512) are calculated, and the results are shown in Fig. 3 (a) and 3 (b), respectively. It is observed that the simulation results of molar volume are in good agreement with the experimental data [36, 49]. The molar volume of the pure [bmim][PF6] is markedly higher than that of the pure [bmim][NO3]. The main reason is that the size of the anion has an effect on the molar volume, i.e., smaller anions yield smaller molar volumes [36]. The molar volumes of two binary systems of [bmim][NO3]-CO2and [bmim][PF6]-CO2decrease with the increase of the mole fraction of CO2because the volumes of the mixtures increase slightly even though a large amount of CO2dissolve in the liquid phase. The molar volume of [bmim][PF6]-CO2is larger than that of [bmim][NO3]-CO2at the same mole fraction of CO2, which should be relevant to the size of anion.
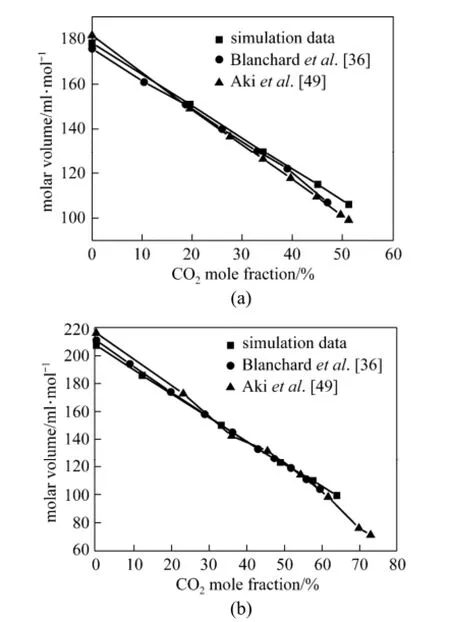
Figure 3 Molar volume of [bmim][NO3]-CO2with the increase of the mole fraction of CO2(a) and molar volume of [bmim][PF6]-CO2with the increase of the mole fraction of CO2(b)
To explain the reason for the low volume expansion of IL-CO2systems with high mole fraction of CO2, Berne et al. [25] have first proposed the hypothesis that some void spaces were inter and intra IL molecules. When CO2dissolve into IL, the small void spaces will be rearranged to form big ones to accommodate more CO2molecules, and thus large volume expansion of IL cannot be observed. The unique characteristic of IL-CO2is beneficial to its industrial applications because the addition of CO2decreased the solvent strength of ILs to a very small extent while the transport properties are improved by increasing diffusion coefficient and decreasing viscosity of ILs. In addition, larger reactors are not needed when using these mixtures as solvents due to its slightly increased volume and the cost of apparatus will be reduced.
3.3 Microstructures
Many researches have reported the unique properties for IL-CO2systems by experimental method [18, 20, 36, 49]. For instance, the viscosity of ILs significantly decreases with addition of CO2and the volumes of the mixtures increase slightly even though a large amount of CO2is dissolved in the liquid phase. The similar results via MD simulation method were obtained as mentioned above. Berne et al. [25] proposed a hypothesis that there are some void spaces inter and intra IL molecules, which can explain these puzzling phenomenon from microscopic view. In order to understand the possible change of microstructures of ILs after the addition of CO2and find out how the microstructures affect their properties, site to site radial distribution functions (RDFs) for the cationanion, H-O, H-F, anion-anion, and CO2-anion were investigated for [bmim][NO3]-CO2and [bmim][PF6]-CO2systems. RDFs were used to evaluate the distance-dependent relative probability of observing at a given site or atom relative to some central site or atom and to reflect the microstructure of the fluids. The microstructures can also be expressed by the coordination numbers (N) of the first solvent shell. That is the average number of specific sites or atoms within a sphere of radius Rsabout some other central site or atom. The value of N in the first solvent shell can be obtained by integration of the RDFs from zero to the Rmin1[Eq. (6)] [51].

where Rmin1refers to the first minimum in g(Rs), ρ is the number density, and g(Rs) is the ratio between local number density and bulk number density of sites or atoms which are in the shell of radius Rsand thickness dRs. Since length of all cubic boxes obtained after simulation are more than 3 nm, the cutoff distance for RDFs analysis is set to 1.5 nm, which is about the half-length of the simulation box. The calculated results of RDFs and N are shown in Figs. 4-9 and discussed as follows, respectively.
3.3.1 Cation-N(NO3)/P(PF6)
The interaction between cation and two types of anion were studied by cation-N/P RDFs and corresponding number integrals (CNIs) of N/P around cation. The results are presented in Figs. 4 and 5. From Fig. 4 (a), it is known that the positions of the first peak and first minimum of RDFs shift slightly in [bmim][PF6]-CO2with the increase of mole fraction of CO2, which is in good agreement with the previous simulation results [24]. The positions of the first peak and first minimum of RDFs for [bmim][PF6]-CO2mixtures at different CO2concentrations are all located at about 0.45 nm and 0.74 nm, respectively. The two positions shift a little for [bmim][NO3]-CO2mixtures at different CO2concentrations, and they are all located at about 0.42 nm and 0.71 nm, respectively, as shown in Fig. 5 (a). Furthermore, the shapes of all the curves are similar and only the peak heights change a little for both RDFs of [bmim][PF6]-CO2and [bmim][NO3]-CO2systems. The slight changes of RDFs demonstrate that there is almost no change of microstructures inside the IL molecules with addition of CO2and some void spaces are inter and intra IL molecules to accommodate CO2molecules. As seen in Fig. 4 (b), the coordination numbers of P atom around cation are 5.6, 5.4, 5.2, 5.0, 4.9 and 4.8 when CO2mole fraction are 0, 0.123, 0.333, 0.490, 0.576 and 0.639, respectively, while for N atom they are also 5.6, 5.5, 5.3, 5.2 and 5.0 when CO2mole fraction are 0, 0.200, 0.342, 0.451 and 0.512, respectively, as shown in Fig. 5 (b). The coordination numbers of both P atom and N atom around cation decrease with the increase of CO2concentrations, implying that the cations and anions move away from each other with introduction of CO2. Thus, the hydrogen bonds interactions between cation and anion are weakened by CO2molecules because the cations and anions connect each other to form a hydrogen-bonded network in ILs [36].

Figure 4 RDFs of cation-P(PF6) at different mole fraction of CO2in the mixtures (a) and CNIs of P around cation (b) (The mass center of the imidazolium ring is taken as the position of the cation) mole fraction/%: ■ 0; ● 12.3; ▲ 33.3; ▼ 49.0;57.6;63.9

Figure 5 RDFs of cation-N(NO3) at different mole fraction of CO2in the mixtures (a) and CNIs of N around cation (b) (The mass center of the imidazolium ring is taken as the position of the cation) mole fraction/%: ■ 0; ● 20.0; ▲ 34.2; ▼ 45.1;51.2
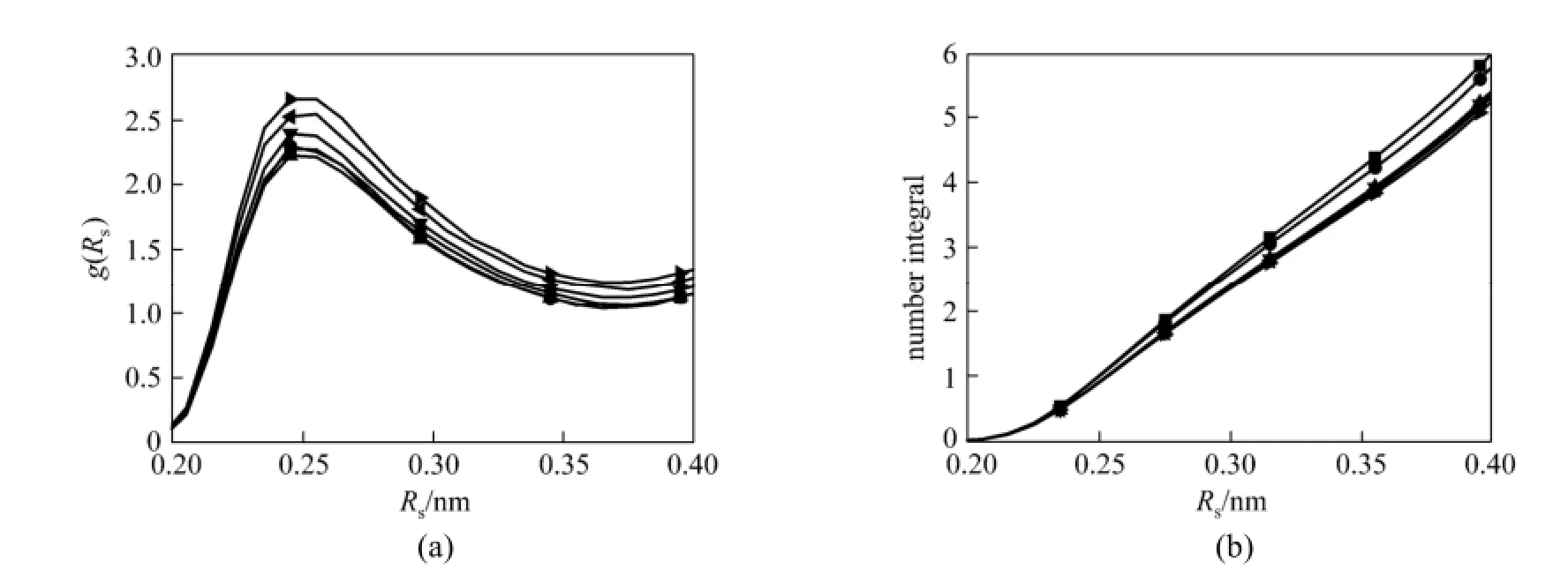
Figure 6 RDFs of H-F(PF6) at different mole fraction of CO2in the [bmim][PF6]-CO2mixtures (a) and CN+Is of F around H atom (b) (The H atom connects to the carbon atom between the two nitrogen atoms in imidazolium ring of [bmim]) mole fraction/%: ■ 0; ● 12.3; ▲ 33.3; ▼ 49.0;57.6;63.9
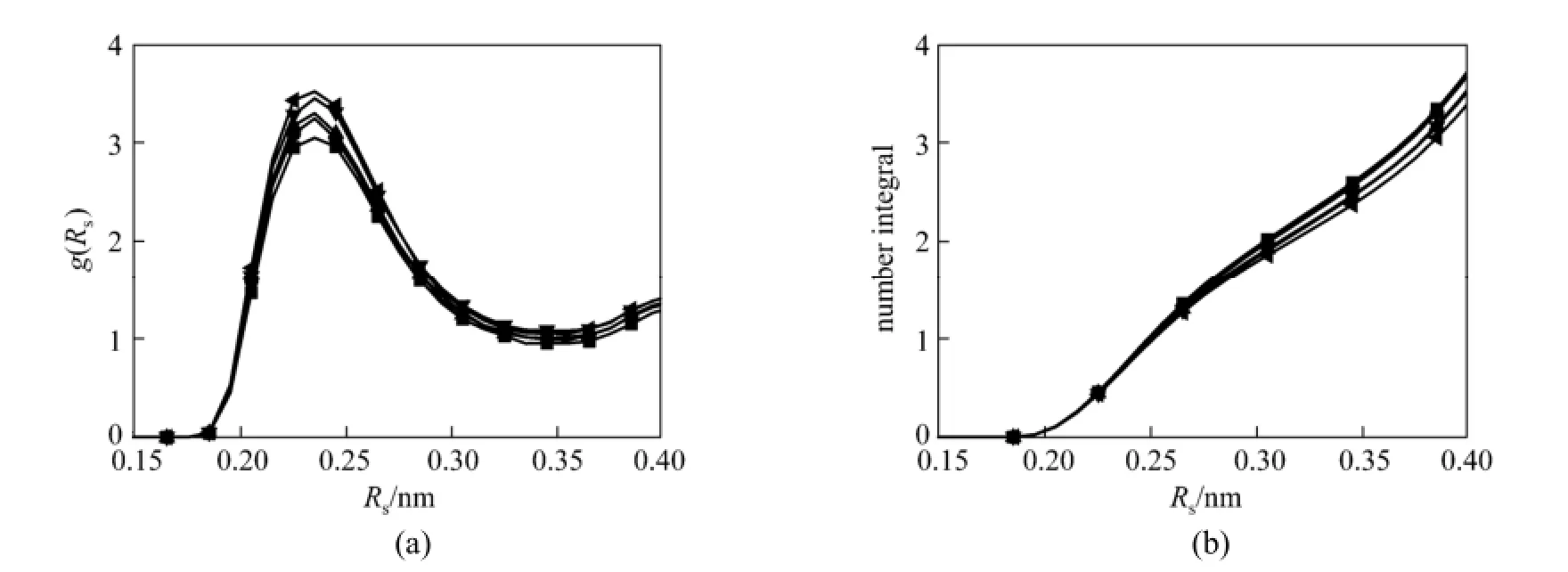
Figure 7 RDFs of H-O(NO3) at different mole fraction of CO2in the [bmim][NO3]-CO2mixtures (a) and CNI+s of O around H atom (b) (The H atom connects to the carbon atom between the two nitrogen atoms in imidazolium ring of [bmim]) mole fraction/%: ■ 0; ● 20.0; ▲ 34.2; ▼ 45.1;51.2
To further describe hydrogen bonds interaction of [bmim][PF6] and [bmim][NO3] ILs, the RDFs of H-F(PF6)/O(NO3) and CNIs of F/O around H atom (H atom connects to the carbon atom between the two nitrogen atoms in imidazolium ring of [bmim]+) are investigated, and the results are shown in Figs. 6 and 7. As it can be seen in Fig. 6 (a), the RDFs of H-F are similar at all mole fraction of CO2. The positions of the first peak and the first minimum are located at 0.25 nm and 0.38 nm, respectively. The position of the first peak is unaltered with increase of mole fraction of CO2because the length of the hydrogen bonds between H atom and F atom is a constant. The RDFs of H-O in Fig. 7 (a) also present the same trend. The positions of the first peak and the first minimum at all mole fraction of CO2are located at 0.24 nm and 0.35 nm, respectively. From Fig. 6 (b), it is known that the coordination numbers of F atom around H atom from 0 to 0.38 nm (the location of the first minimum) are 5.2, 5.0, 4.7, 4.6, 4.5 and 4.4 when CO2mole fraction are 0, 0.123, 0.333, 0.490, 0.576 and 0.639, respectively, while for O atom around H atom they are 2.7, 2.6, 2.5, 2.5 and 2.4 when CO2mole fraction are 0, 0.200, 0.342, 0.451 and 0.512, respectively, as shown in Fig. 7 (b). The coordination numbers of both F atom and O atom around H atom decrease with the increase of mole fraction of CO2, which implies that the hydrogen bonds interaction is weakened by addition of CO2molecules. Hydrogen bonds interaction between the molecules can affect the transport properties, such as the diffusion coefficient and viscosity [52] and it also limits the molecular random movement of ILs. The stronger the hydrogen bonds interaction between cation and anion is, the higher the viscosity of ILs is. Thus, the addition of CO2molecules can increase the diffusion coefficient and decrease the viscosity of ILs.
3.3.2 N(NO3)-N(NO3)/P(PF6)-P(PF6)
Figure 8 presents the RDFs of N(NO3)-N(NO3)/ P(PF6)-P(PF6) and CNIs. The main purpose is to investigate the influence of CO2molecules on interactions among anions. From Fig. 8 (a), it can be seen that the position of first peak in RDFs of P-P varies significantly with increase of mole fraction of CO2. The position of the first peak for RDFs of P-P shifts from 0.66 nm in the case of pure [bmim][PF6] to a value of 0.78 nm when CO2mole fraction is 0.639. And the position of the first minimum is located at 1.12 nm at all mole fraction of CO2. Moreover, a shoulder peak appears near the first peak at 0 mole fraction of CO2, whereas the location of the first minimum keeps almost unchanged. Balasubramanian et al. [24] have also presented that the interaction between [PF6]−-[PF6]−is significantly affected by adding CO2based on RDFs. But for RDFs of N-N, the position of first peak changes a little. The positions of the first peak are located at about 0.68 nm and 0.72 nm when CO2mole fraction are 0 and 0.512, respectively, and the position of the first minimum is located at 1.04 nm at all mole fraction of CO2. Obviously, the position of the first peak of P-P RDFs changes more than that of N-N, indicating that the effect of CO2on the interaction between [PF6]−-[PF6]−is stronger than that of [NO3]−-[NO3]−.
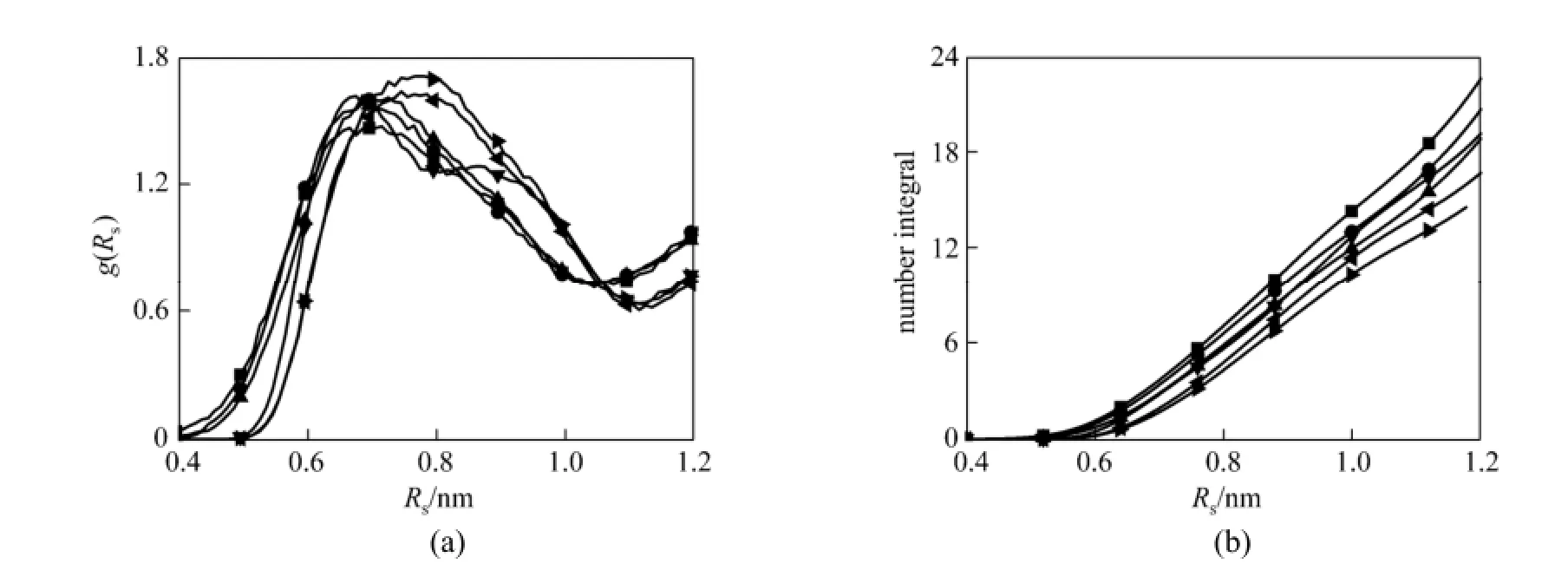
Figure 8 RDFs of N(NO3)-N(NO3) and P(PF6)-P(PF6) at different mole fraction of CO2(a) and CNIs of N around N and P around P (b) mole fraction/%: ■ 0, N-N; ● 34.2, N-N; ▲ 51.2, N-N; ▼ 0, P-P;33.3, P-P;63.9, P-P
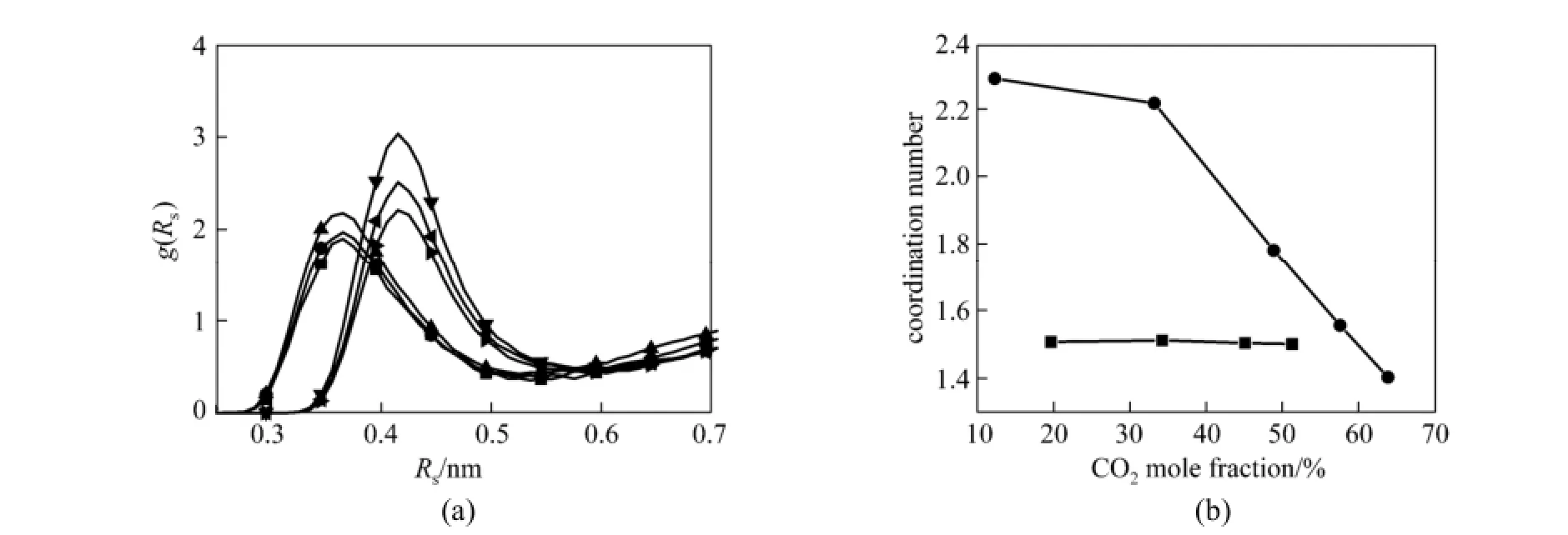
Figure 9 RDFs of C(CO2)-N(NO3) and C(CO2)-P(PF6) at different mole fraction of CO2(a) and CNIs of N and P around C (b) mole fraction/%: ■ 20.0, C-H; ● 34.2, C-N; ▲ 51.2, C-N; ▼ 33.3, C-P;49.0, C-P;63.9, C-P
As seen in Fig. 8 (b), the coordination numbers of N(NO3) atom around N(NO3) atom are 15.6, 14.2, and 13.0 when CO2mole fraction are 0, 0.342 and 0.512, respectively. The coordination numbers of P(PF6) atom around P(PF6) atom are 16.4, 15.8 and 13.1 when CO2mole fraction are 0, 0.333 and 0.639, respectively. The decrease of anion-anion coordination numbers is due to the anions moving away from each other with introduction of CO2. For [bmim][PF6]-CO2and [bmim][NO3]-CO2systems, both coordination numbers of anion around anion decrease with the addition of CO2, which implies that the interaction of anion-anion is significantly weakened by the CO2molecules.
3.3.3 C(CO2)-N(NO3)/C(CO2)-P(PF6)
The interaction between anions and CO2is investigated by RDFs of C(CO2)-N(NO3) and C(CO2)-P(PF6) and CNIs, and the results are presented in Fig. 9. The RDFs of C(CO2)-N(NO3) and C(CO2)-P(PF6) are presented in Fig. 9 (a). It can be seen that the positions of the first peak and the first minimum of RDFs of C(CO2)-N(NO3) and C(CO2)-P(PF6) keep almost unchanged with the increase of mole fraction of CO2. For [bmim][NO3]-CO2systems, the first peak and the first minimum are presented at 0.36 nm and 0.53 nm, respectively. For [bmim][PF6]-CO2systems, they are located at 0.41 nm and 0.58 nm, respectively. The radius of the first coordination shell is the distance from zero to the first minimum of the RDFs. Thus, the radius of the first coordination shell for C(CO2)-N(NO3) RDFs are smaller than those of C(CO2)-P(PF6). Obviously, the peak heights of C(CO2)-P(PF6) RDFs decrease with the increase of the mole fraction of CO2, while those of C(CO2)-N(NO3) RDFs increase. This strange phenomenon may be attributed to the different structures and properties of ILs. Brennecke et al. [53] have also proposed that the type of anion has great influence on the structures and properties of ILs.
The coordination numbers of P(PF6)/N(NO3) around CO2are presented in Fig. 9 (b). From this figure, it is found that the coordination numbers of P(PF6) atom around C(CO2) atom are 2.2, 1.8 and 1.4 when CO2mole fraction are 0.333, 0.490 and 0.639, respectively. But for [bmim][NO3]-CO2mixtures, the coordination numbers of N(NO3) atom around C(CO2) atom are about 1.5 at all mole fraction of CO2. From Fig. 9 (b), it can be also seen that there is an obvious decrease of coordination numbers of P atom around C atom, especially when CO2mole fraction is more than 0.333, while the coordination numbers curve for C(CO2)-N(NO3) is almost flat. These differences havea certain relationship with the structure of ILs. When CO2mole fraction is less than 0.333, the added CO2occupies the void spaces of IL molecules and has a little influence on the structure of ILs. With continuous addition of CO2molecules, the void spaces of IL molecules are filled with CO2molecules and then the structure of IL molecules has to significantly change in order to accommodate more CO2molecules. In addition, it can be seen that when CO2mole fraction ranges from 0 to 0.333, the coordination numbers of P(PF6) atom around P(PF6) atom decrease by 0.8, while the coordination numbers of P(PF6) atom around P(PF6) atom decrease by 2.7 with CO2mole fraction from 0.333 to 0.639 as shown in Fig. 8 (b). This result demonstrates that the smaller probability of P atom appears in a sphere of radius r and essentially indicates that the anions move further away from each other when CO2mole fraction is higher than 0.333. Therefore, an obvious decrease of coordination numbers of P atom around C atom is observed, especially when CO2mole fraction is higher than 0.333. Furthermore, the coordination number of P atom around C atom is larger than that of N atom around C atom at the same mole fraction of CO2(from 0 to 0.512). It can be explained by that the interaction between CO2and [PF6]−is stronger than that between CO2and [NO3]−, and thus the effect of adding CO2to [bmim][PF6] is stronger than that of [bmim][NO3]. This also explains that [bmim][NO3] can be used to form thermodynamics stable scCO2reverse micelles as the polarity molecule, whereas [bmim][PF6] cannot, as proposed by Chandran et al [54]. Thus, it is possible to design and tune the type of anion of ILs that have weak interaction with CO2but strong interaction with headgroup of surfactant to form thermodynamics stable of ionic liquid-in-carbon dioxide (IL-in-CO2) reverse micelles with the help of MD simulation.
4 CONCLUSIONS
Molecular dynamics simulation has been carried out on the two binary systems of [bmim][PF6]-CO2and [bmim][NO3]-CO2at the different mole fraction of CO2. The diffusion coefficient of ILs increases with the increase of mole fraction of CO2. When CO2mole fraction is 0.512, the diffusion coefficient of [bmim][NO3] for [bmim][NO3]-CO2system is 4.5 times as high as that of the pure [bmim][NO3], while that of [bmim][PF6] for [bmim][PF6]-CO2system with CO2mole fraction of 0.639 is 7.9 times as high as that of the pure [bmim][PF6]. The viscosity of ILs decreases with the increase of mole fraction of CO2. The viscosity of the pure [bmim][NO3] is 3.1 times as high as that of [bmim][NO3]-CO2system with CO2mole fraction of 0.512, while the viscosity of the pure [bmim][PF6] is 5.4 times as high as that of [bmim][PF6]-CO2system with CO2mole fraction of 0.639.
The molar volumes decrease with the increase of the mole fraction of CO2of the two binary systems and the volume expansion of ILs is very small with addition of CO2and less than 35% for the two binary systems, which is quite different to CO2expanded organic solvents. This is consistent with the experimental results in the previously reports and the interesting phenomenon is mainly due to some void spaces existing inter and intra IL molecules.
For the two binary systems, site to site RDFs and the CNIs of the cation-anion, H-O, H-F, anion-anion, and CO2-anion are investigated. The simulating results indicate that the microstructures of ILs have been changed a little, but the properties of ILs have been changed a lot after the addition of CO2. The main conclusions are as follows: (1) as the cations and anions moving away from each other with introduction of CO2, the structure of ILs is changed and the hydrogen bonds interaction between cation-anion are also weakened by the addition of CO2, which leads to decrease of the viscosity and increase of the diffusion coefficient of ILs. (2) the interaction between CO2and [PF6]−is stronger than that between CO2and [NO3]−, which explains that [bmim][NO3] can be used to form thermodynamics stable scCO2reverse micelles, whereas [bmim][PF6] cannot. This provides a helpful proof to design new ILs to form thermodynamics stable of IL-in-CO2reverse micelles. (3) the type of anion has a great influence on the properties of ILs by comparing [bmim][PF6] with [bmim][NO3].
NOMENCLATURE
D diffusion coefficient, m2·s−1
dRsthickness, m
g(Rs) the ratio between local number density and bulk number density of sites or atoms
K force constant
N number
n periodicity
p pressure, MPa
q charges, C
r length, m
Δr distance, m
T temperature, K
t time, ns
U potential energy, kJ·mol−1
V volume, m3
ΔVrvolume expansion ratio, %
γ equilibrium dihedral angle, rad
ε traditional well-depth
θ angle, rad
ρ number density, m−3
σ distance, m
ϕ dihedral angle, rad
Subscripts
b bonds
c critical
i, j atoms
L total volume of the liquid mixture at a given temperature and pressure
REFERENCES
1 Blanchard, L.A., Hancu, D., Beckman, E.J., Brennecke, J.F., “Greenprocessing using ionic liquids and CO2”, Nature, 399 (6731), 28-29 (1999).
2 Lin, I.H., Tan, C.S., “Diffusion of benzonitrile in CO2-expanded ethanol”, J. Chem. Eng. Data, 53 (8), 1886-1891 (2008).
3 Sun, H.W., “Ionic liquids: Progress and prospective”, Chin. J. Chem. Eng., 13 (6), 830-834 (2005).
4 Earle, M.J., Esperanca, J., Gilea, M.A., Lopes, J.N.C., Rebelo, L.P.N., Magee, J.W., Gilea, K.R., Seddon, M.A., Widegren, J.A.,“The distillation and volatility of ionic liquids”, Nature, 439 (7078), 831-834 (2006).
5 Wasserscheid, P., “Chemistry—Volatile times for ionic liquids”, Nature, 439 (7078), 797-797 (2006).
6 Zhao, Z., Dong, H., Zhang, X., “The research progress of CO2capture with ionic liquids”, Chin. J. Chem. Eng., 20 (1), 120-129 (2012).
7 Earle, M.J., Seddon, K.R., “Ionic liquids. Green solvents for the future”, Pure Appl. Chem., 72 (7), 1391-1398 (2000).
8 Fang, S., Zhang, Z., Jin, Y., Yang, L., Hirano, S., Tachibana, K., Katayama, S., “New functionalized ionic liquids based on pyrrolidinium and piperidinium cations with two ether groups as electrolytes for lithium battery”, J. Power Sources, 196 (13), 5637-5644 (2011).
9 Blanchard, L.A., Brennecke, J.F., “Recovery of organic products from ionic liquids using supercritical carbon dioxide”, Ind. Eng. Chem. Res., 40 (1), 287-292 (2000).
10 Wang, W., Yin, J., “CO2/ionic liquids phase Behaviors and its applications for reaction and separation”, Prog. Chem., 20 (4), 441-449 (2008). (in Chinese)
11 Shiflett, M.B., Yokozeki, A., “Solubilities and diffusivities of carbon dioxide in ionic liquids: [bmim][PF6] and [bmim][BF4]”, Ind. Eng. Chem. Res., 44 (12), 4453-4464 (2005).
12 Lu, J., Liotta, C.L., Eckert, C.A., “Spectroscopically probing microscopic solvent properties of room-temperature ionic liquids with the addition of carbon dioxide”, J. Phys. Chem. A, 107 (19), 3995-4000 (2003).
13 Tomida, D., Kumagai, A., Qiao, K., Yokoyama, C., “Viscosity of 1-butyl-3-methylimidazolium hexafluorophosphate + CO2mixture”, J. Chem. Eng. Data, 52 (5), 1638-1640 (2007).
14 Babarao, R., Dai, S., Jiang, D.E., “Understanding the high solubility of CO2in an ionic liquid with the tetracyanoborate anion”, J. Phys. Chem. B, 115 (32), 9789-9794 (2011).
15 Perez-Blanco, M.E., Maginn, E.J., “Molecular dynamics simulations of CO2at an ionic liquid interface: Adsorption, ordering, and interfacial crossing”, J. Phys. Chem. B, 114 (36), 11827-11837 (2010).
16 Wang, Y., Pan, H., Li, H., Wang, C., “Force field of the TMGL ionic liquid and the solubility of SO2and CO2in the TMGL from molecular dynamics simulation”, J. Phys. Chem. B, 111 (35), 10461-10467 (2007).
17 Wang, W.B., Yin, J.Z., Sun, L.H., Feng, E., “Molecular dynamics simulation of thermodynamic properties for CO2/ionic liquid systems”, Acta Phys. Chim. Sin., 25 (11), 2291-2295 (2009). (in Chinese)
18 Kazarian, S.G., Briscoe, B.J., Welton, T., “Combining ionic liquids and supercritical fluids: in situ ATR-IR study of CO2dissolved in two ionic liquids at high pressures”, Chem. Commun., 36 (20), 2047-2048 (2000).
19 Fredlake, C.P., Muldoon, M.J., Aki, S., Welton, T., Brennecke, J.F., “Solvent strength of ionic liquid/CO2mixtures”, Phys. Chem. Chem. Phys., 6 (13), 3280-3285 (2004).
20 Kanakubo, M., Umecky, T., Hiejima, Y., Aizawa, T., Nanjo, H., Kameda, Y., “Solution structures of 1-butyl-3-methylimidazolium hexanuorophosphate ionic liquid saturated with CO2: Experimental evidence of specific anion-CO2interaction”, J. Phys. Chem. B, 109 (29), 13847-13850 (2005).
21 Hou, Y., Baltus, R.E., “Experimental measurement of the solubility and diffusivity of CO2in room temperature ionic liquids using a transient thin-liquid-film method”, Ind. Eng. Chem. Res., 46 (24), 8166-8175 (2007).
22 Shim, Y., Kim, H.J., “MD study of solvation in the mixture of a room temperature ionic liquid and CO2”, J. Phys. Chem. B, 114 (31), 10160-10170 (2010).
23 Bhargava, B.L., Balasubramanian, S., “Insights into the structure and dynamics of a room temperature ionic liquid: Ab initio molecular dynamics simulation studies of 1-n-butyl-3-methylimidazolium hexafluorophosphate ([bmim][PF6]) and the [bmim][PF6]-CO2mixture”, J. Phys. Chem. B, 111 (17), 4477-4487 (2007).
24 Bhargava, B.L., Krishna, A.C., Balasubramanian, S., “Molecular dynamics simulation studies of CO2-[bmim][PF6] solutions: Effect of CO2concentration”, AIChE J., 54 (11), 2971-2978 (2008).
25 Huang, X., Margulis, C.J., Li, Y., Berne, B.J., “Why is the partial molar volume of CO2so small when dissolved in a room temperature ionic liquid? Structure and dynamics of CO2dissolved in [Bmim+][PF6−]”, J. Am. Chem. Soc., 127 (50), 17842-17851 (2005).
26 Cadena, C., Anthony, J.L., Shah, J.K., Morrow, T.I., Brennecke, J.F., Maginn, E.J., “Why is CO2so soluble in imidazolium-based ionic liquids?”, J. Am. Chem. Soc., 126 (16), 5300-5308 (2002).
27 Canongia Lopes, J.N., Deschamps, J., Pádua, A.A.H., “Modeling ionic liquids using a systematic all-atom force field”, J. Phys. Chem. B, 108 (6), 2038-2047 (2004).
28 Liu, Z., Huang, S., Wang, W., “A refined force field for molecular simulation of imidazolium-based ionic liquids”, J. Phys. Chem. B, 108 (34), 12978-12989 (2004).
29 Micaelo, N.M., Baptista, A.M., Soares, C.M., “Parametrization of 1-butyl-3- methylimidazolium hexafluorophosphate/nitrate ionic liquid for the GROMOS force field”, J. Phys. Chem. B, 110 (29), 14444-14451 (2006).
30 Cornell, W.D., Cieplak, P., Bayly, C.I., Gould, I.R., Merz, K.M., Ferguson, D.M., Spellmeyer, D.C., Fox, T., Caldwell, J.W., Kollman, P.A., “A second generation force field for the simulation of proteins, nucleic acids, and organic molecules”, J. Am. Chem. Soc., 117 (19), 5179-5197 (1995).
31 Yan, T., Burnham, C.J., Del Pópolo, M.G., Voth, G.A., “Molecular dynamics simulation of ionic liquids: The effect of electronic polarizability”, J. Phys. Chem. B, 108 (32), 11877-11881 (2004).
32 Houndonougbo, Y., Jin, H., Rajagopalan, B., Wong, K., Kuczera, K., Subramaniam, B., Laird, B., “Phase equilibria in carbon dioxide expanded solvents: Experiments and molecular simulations”, J. Phys. Chem. B, 110 (26), 13195-13202 (2006).
33 Qin, Y., Yang, X., Zhu, Y., Ping, J., “Molecular dynamics simulation of interaction between supercritical CO2fluid and modified silica surfaces”, J. Phys. Chem. C, 112 (33), 12815-12824 (2008).
34 Senapati, S., Berkowitz, M.L., “Molecular dynamics simulation studies of polyether and perfluoropolyether surfactant based reverse micelles in supercritical carbon dioxide”, J. Phys. Chem. B, 107 (47), 12906-12916 (2003).
35 Liu, Z., Wu, W., Han, B., Dong, Z., Zhao, G., Wang, J., Jiang, T., Yang, G., “Study on the phase behaviors, viscosities, and thermodynamic properties of CO2/[C4mim][PF6]/methanol system at elevated pressures”, Chem. Eur. J., 9 (16), 3897-3903 (2003).
36 Blanchard, L.A., Gu, Z., Brennecke, J.F., “High-pressure phase behavior of ionic liquid/CO2systems”, J. Phys. Chem. B, 105 (12), 2437-2444 (2001).
37 Berendsen, H.J.C., Postma, J.P.M., Van Gunsteren, W.F., DiNola, A., Haak, J.R., “Molecular dynamics with coupling to an external bath”, J. Chem. Phys., 81 (8), 3684-3690 (1984).
38 Essmann, U., Perera, L., Berkowitz, M.L., Darden, T., Lee, H., Pedersen, L.G., “A smooth particle mesh Ewald method”, J. Chem. Phys., 103 (19), 8577-8593 (1995).
39 Dong, K., Zhang, S., Wang, D., Yao, X., “Hydrogen bonds in imidazolium ionic liquids”, J. Phys. Chem. A, 110 (31), 9775-9782 (2006).
40 Morgan, D., Ferguson, L., Scovazzo, P., “Diffusivities of gases in room temperature ionic liquids: Data and correlations obtained using a lag-time technique”, Ind. Eng. Chem. Res., 44 (13), 4815-4823 (2005).
41 Rapaport, D.C., The Art of Molecular Dynamics Simulation, 2ndedition, Cambridge University Press, Cambridge (2004).
42 Shiflett, M.B., Yokozeki, A., “Solubility and diffusivity of hydrofluorocarbons in room temperature ionic liquids”, AIChE J., 52 (3), 1205-1219 (2006).
43 Kim, D.H., Baek, I.H., Hong, S.U., Lee, H.K., “Study on immobilized liquid membrane using ionic liquid and PVDF hollow fiber as a support for CO2/N2separation”, J. Membr. Sci., 372 (1/2), 346-354 (2011).
44 Iarikov, D.D., Hacarlioglu, P., Oyama, S.T., “Supported room temperature ionic liquid membranes for CO2/CH4separation”, Chem. Eng. J., 166 (1), 401-406 (2011).
45 Knez, Z., Weidner, E., “Particles formation and particle design using supercritical fluids”, Curr. Opin. Solid State Mater. Sci., 7 (4/5), 353-361 (2003).
46 Shariati, A., Peters, C.J., “Recent developments in particle design using supercritical fluids”, Curr. Opin. Solid State Mater. Sci., 7 (4/5), 371-383 (2003).
47 de la Fuente Badilla, J.C., Peters, C.J., de Swaan Arons, J., “Volume expansion in relation to the gas—Antisolvent process”, Fluid Phase Equilib., 17 (1), 13-23 (2000).
48 Gallagher, P.M., Coffey, M.P., Krukonis, V.J., Klasutis, N., “Gas antisolvent recrystallization: New process to recrystallize compounds insoluble in supercritical fluids”, In: Supercritical Fluid Science and Technology, American Chemical Society, USA, 406 (22), 334-354 (1989).
49 Aki, S.N.V.K., Mellein, B.R., Saurer, E.M., Brennecke, J.F.,“High-pressure phase behavior of carbon dioxide with imidazoliumbased ionic liquids”, J. Phys. Chem. B, 108 (52), 20355-20365 (2004).
50 Kordikowski, A., Schenk, A.P., Van Nielen, R.M., Peters, C.J., “Volume expansions and vapor-liquid equilibria of binary mixtures of a variety of polar solvents and certain near-critical solvents”, J. Supercrit. Fluids., 8 (3), 205-216 (1995).
51 Liu, X., Zhou, G., Zhang, S., Wu, G., Yu, G., “Molecular simulation of guanidinium-based ionic liquids”, J. Phys. Chem. B, 111 (20), 5658-5668 (2007).
52 Fioroni, M., Burger, K., Mark, A.E., Roccatano, D., “A new 2,2,2-trifluoroethanol model for molecular dynamics simulations”, J. Phys. Chem. B, 104 (51), 12347-12354 (2000).
53 Fredlake, C.P., Crosthwaite, J.M., Hert, D.G., Aki, S.N.V.K., Brennecke, J.F., “Thermophysical properties of imidazolium-based ionic liquids”, J. Chem. Eng. Data, 49 (4), 954-964 (2002).
54 Chandran, A., Prakash, K., Senapati, S., “Self-assembled inverted micelles stabilize ionic liquid domains in supercritical CO2”, J. Am. Chem. Soc., 132 (35), 12511-12516 (2010).
Received 2012-11-15, accepted 2013-01-25.
* Supported by the National Natural Science Foundation of China (20976026, 20976028) and the Natural Science Foundation of Liaoning Province (20102030, 20031072).
** To whom correspondence should be addressed. E-mail: jzyin@dlut.edu.cn
猜你喜欢
杂志排行

Chinese Journal of Chemical Engineering的其它文章
- Effects of Solvent and Impurities on Crystal Morphology of Zinc Lactate Trihydrate*
- Numerical Simulation of Oxy-coal Combustion for a Swirl Burner with EDC Model*
- Performance of EDAB-HCl Acid Blended System as Fracturing Fluids in Oil Fields*
- Kinetic and Thermodynamic Studies of Acid Scarlet 3R Adsorption onto Low-cost Adsorbent Developed from Sludge and Straw*
- Large-eddy Simulation of Ethanol Spray-Air Combustion and Its Experimental Validation*
- Synthesis of Sub-micrometer Lithium Iron Phosphate Particles for Lithium Ion Battery by Using Supercritical Hydrothermal Method