取代芳香族冠醚配合物的合成、晶体结构及其与DNA相互作用的研究
2014-01-02牛茂斐常文明牛美菊
牛茂斐 常文明 牛美菊
(聊城大学化学化工学院,聊城 252059)
冠醚化学作为一门植根深远的新兴边缘学科,现已渗透到生命科学、材料科学、环境科学、医药学等重要领域[1-3],并已取得了令人瞩目的成就,所以进一步研究新型冠醚化合物及其配合物的合成方法、结构和性质具有十分重要的意义。对于芳香族取代冠醚因有更佳的光谱性质和更易功能化等特点,它们的分子设计和合成尤为重要。文献对4-位取代芳香族冠醚及其配合物的合成和晶体结构已有一些报道[4-6],但对3-位取代芳香族冠醚及其配合物的相关研究内容较少[7],尤其是对冠醚配合物与DNA作用等性质研究鲜见文献报道[8]。本文用简单的方法合成了冠醚3,3′-二甲基二苯并-18-冠-6,以合成的冠醚化合物为配体,分别与氯化镍、氯化钯及异丁二腈烯二硫醇钾[K2(i-mnt)]反应,得到了镍(Ⅱ)冠醚配合物1和钯(Ⅱ)冠醚配合物2,通过元素分析、红外光谱、核磁共振氢谱、紫外光谱等方法对其结构进行表征,用X-射线单晶衍射测定了配合物1和配合物2的晶体结构。并通过紫外、荧光和圆二色光谱等分析法,初步研究了配合物与小牛胸腺DNA(CT-DNA)的相互作用性质。
1 实验部分
1.1 仪器与试剂
PE-2400Ⅱ型元素分析仪,Nicolet-5700型红外光谱仪(KBr压片),Mercury Plus-400型核磁共振仪,Bruker Smart-1000 CD型X-射线单晶衍射仪,HP-8453 A型紫外-可见分阵列二极管分光光度计,HP 6890 GC/5973 MSD气-质联用分析仪,附1701 BA-B.01.00 ChemStation软件,Perkin-Elmer公司热分析仪。
溴化乙锭(EB)和小牛胸腺DNA(CT-DNA)购自华美生物工程公司,2,2′-二氯二甘醇醚参考文献[9]合成,K2(i-mnt)参考文献[10]合成,其它均为市售的分析纯试剂。
1.2 3,3′-二甲基二苯并-18-冠-6 的合成
在N2保护下将3.2 g(0.08 mol)NaOH及9.92 g(0.08 mol)3-甲基邻苯二酚加入盛有40 mL异戊醇和DMF 100 mL的四口烧瓶中,加热搅拌回流,5 min内滴加完6 g(0.04 mol)2,2′-二氯二甘醇醚,反应1.5 h后,再次加入3.2 g(0.08 mol)NaOH和6 g(0.04 mol)2,2′-二氯二甘醇醚,继续反应 2 h,冷至室温,减压蒸出溶剂得褐色粘稠物,用氯仿溶解,加入少量中性氧化铝粉末风干,用正庚烷热萃取,萃取液冷冻析出7.3 g产品,产品收率47.04%,m.p.74~77℃。UVVis(CH3OH)λmax:276 nm。1H NMR (CDCl3,400 MHz)δ H:6.70~6.93 (m,6H,Ar-H),3.89~4.20(m,16H,4×OCH2-CH2O),2.68(s,6H,CH3)。IR(KBr,cm-1):3 040(Ar-H),2 920,2 878(C-H),1 587,1 486,1 454(C=C),1 381 (CH3),1 271,1 097(Ar-O-CH2),1 133(CH2-O-CH2),770,748(1,2,3-pehnyl)。 MS(m/z):388(M+),150,136。 元素分析 C22H28O6的计算值 (%):C 68.04, H 7.22;实测值(%):C 68.07,H 7.18。
1.3 冠醚配合物1的合成
在50 mL圆底烧瓶中加入0.059 g(0.025 mol)NiCl2·6H2O,0.109 g(0.05 mol)K2(i-mnt)和 10 mL 水,搅拌1 h后滴加0.388 g(0.1 mol)3,3′-二甲基二苯并-18-冠-6的二氯乙烷溶液10 mL,继续搅拌2 h,分出有机相,有机相加乙醚培养晶体,1周后得适合于X-射线单晶结构分析的黑绿色块状晶体。收率73.62%,m.p.>300 ℃。 UV-Vis(CH3OH)λmax:265 nm。1H NMR(CDCl3,400 MHz)δ H:6.77~7.26(m,6H,Ar-H),3.92~4.26(m,16H,4×OCH2-CH2O),2.26(s,6H,CH3)。IR(KBr,cm-1):2 925,2 881(C-H),2 202(C≡N),1 588,1 476,1 455(C=C),1 381(CH3), 1 267,1 089(Ar-O-CH2),1 124(CH2-O-CH2),771,746 (1,2,3-pehnyl)。 元 素 分 析C58H68K2N4NiO14S4的计算值 (%):C 53.16,H 5.23, N 4.28;实测值(%):C 53.21,H 5.19,N 4.24。
1.4 冠醚配合物2的合成
在50 mL圆底烧瓶中加入0.044 g(0.025 mol)PdCl2,0.109 g(0.05 mol)K2(i-mnt)和 10 mL 水,搅拌1 h 后滴加 0.388 g(0.1 mol)3,3′-二甲基二苯并-18-冠-6的二氯乙烷溶液10 mL,继续搅拌3 h,过滤,沉淀用DMSO和丙酮混合溶剂溶解,2周后得到用于X-射线单晶结构分析的褐黄色块状晶体。收率68.54%,m.p.262~264 ℃。 UV-Vis(CH3OH)λmax:268 nm。1H NMR (CDCl3,400 MHz)δ H:6.80~7.02(m,6H,Ar-H),3.90~4.21 (m,16H,4×OCH2-CH2O),2.26(s,6H,CH3)。IR(KBr,cm-1):2 941,2 880(C-H),2 204(C≡N),1 587,1 485,1 453(C=C),1 382(CH3),1 268,1 093(Ar-O-CH2),1 123(CH2-O-CH2),769,746(1,2,3-pehnyl)。元素分析 C56H68K2N4PdO14S6的计算值 (%):C 48.11,H 4.90,N 4.01; 实 测 值 (%):C 48.14,H 4.86,N 4.01。
1.5 晶体结构的测定
分别选取 0.49 mm×0.43 mm×0.32 mm (1)和0.45 mm×0.42mm×0.40 mm(2)大小的单晶置于Bruker Smart-1000CCD型单晶衍射仪上,用石墨单色化的 Mo Kα(λ=0.071 073 nm)辐射为光源,在298(2)K温度下,分别收集衍射数据。全部强度数据均经Lp因子和经验吸收校正。晶体结构解析和结构精修均采用SHELXTL-97软件[11]完成。结构由直接法解出,其余的非氢原子坐标在以后的数轮差值Fourier合成中陆续确定。对全部非氢原子的坐标及各向异性参数进行了全矩阵最小二乘法修正,氢原子坐标由理论计算确定。配合物的晶体数据列于表1,主要键长和键角列于表2。
CCDC:922022,1;1000767,2.
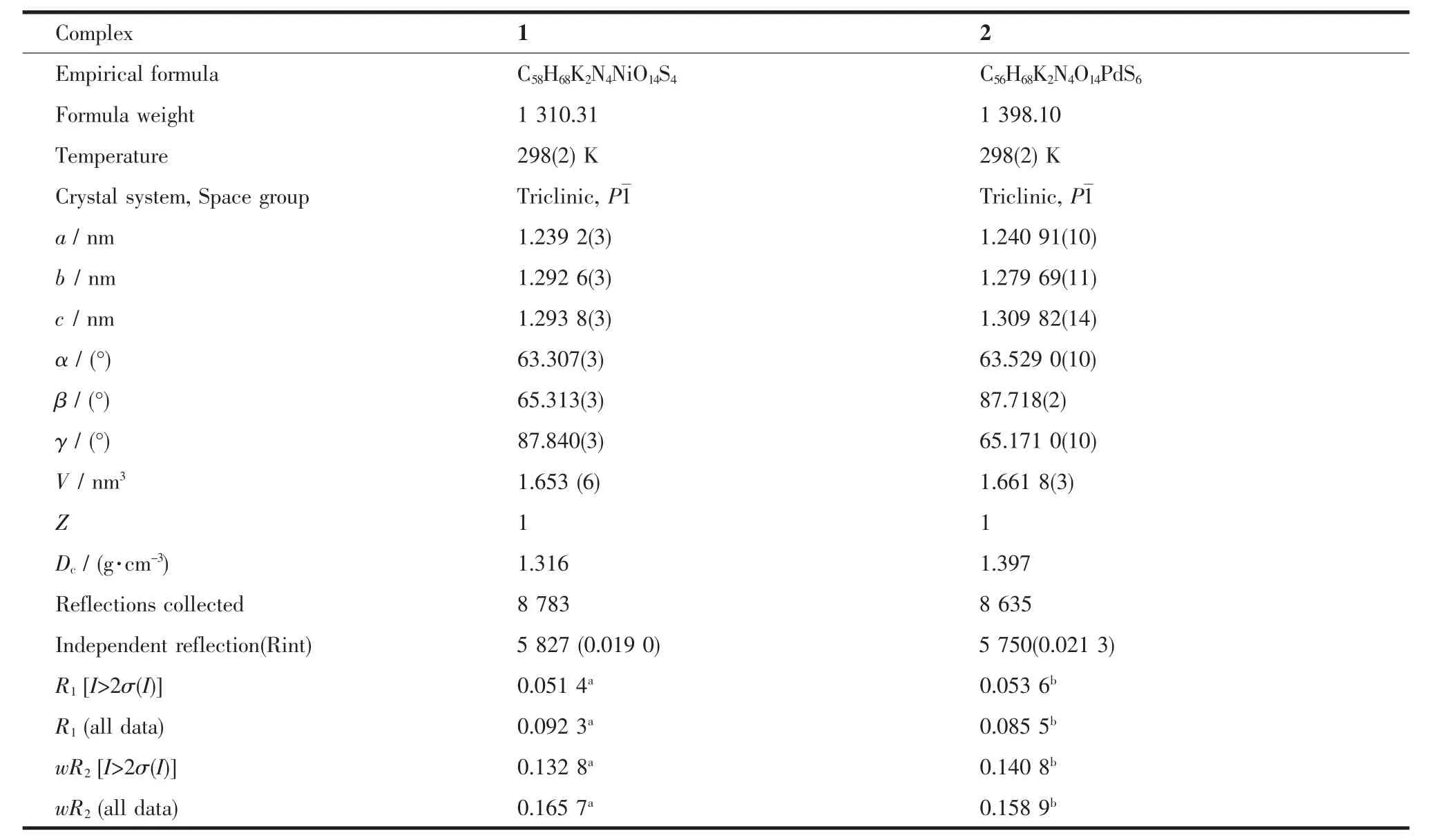
表1 配合物的晶体学数据Table 1 Crystal data and structure refinement of complexes 1 and 2
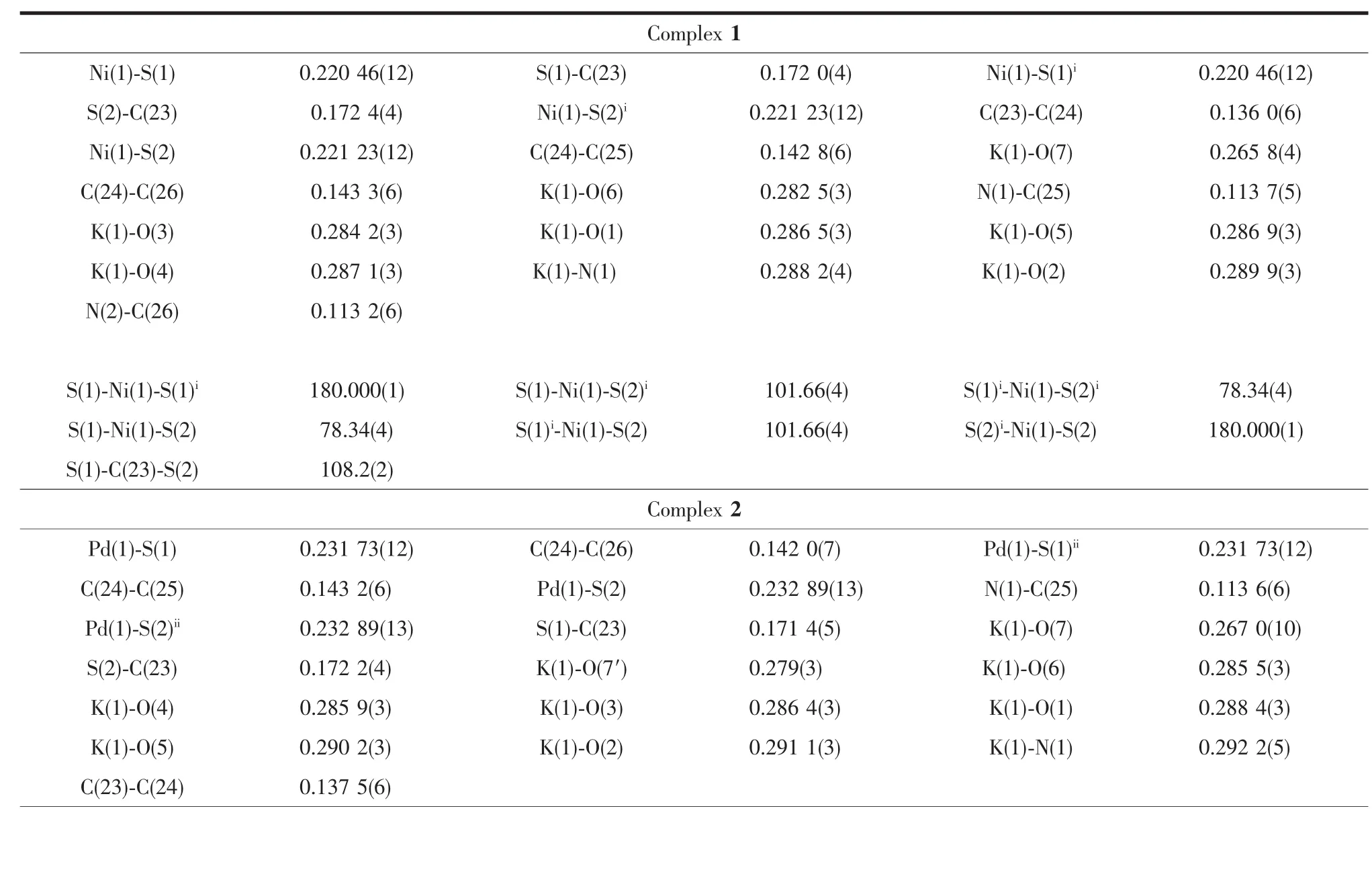
表2 部分键长(nm)和键角(°)Table 2 Selected bond lengths(nm)and angles(°)for complexes 1 and 2

续表2
2 结果与讨论
2.1 合成方法
由于空间阻碍及电子效应等因素的影响,合成3-位取代的芳香族冠醚比较困难,我们选用异戊醇和DMF代替传统合成法中的低碳醇作溶剂,使反应温度提高,反应速度加快,并且DMF是非质子极性溶剂,降低了3-甲基邻苯二酚钠阴离子的溶剂化,从而提高了反应活性,使反应时间缩短到3.5 h。另外,在冠醚合成中产物分离往往是一个棘手的问题,我们直接选用中性氧化铝粉末吸附、风干、热萃取、冷冻,可得纯度较高的产品。该合成方法无酸洗、水洗、溶剂洗、层析等过程,所以具有后处理简单、无污染、反应时间短、产品收率高等优点。
2.2 波 谱
紫外光谱:冠醚配体和配合物均有苯环的特征吸收谱带B带,3类化合物相对于甲苯[UV-Vis(CH3OH)λmax:261 nm][12]的B带吸收均向红移,这是因为取代芳香族冠醚共轭效应的存在,使紫外吸收向长波移动。配体与配合物的吸收位置不同,也发生了位移,说明形成了冠醚配合物。
红外光谱:冠醚配体和配合物均存在苯环、醚氧键及甲基的特征吸收峰,而配合物还在2 202、2 204 cm-1处有C≡N的强吸收峰。对于ν(C-O-C)吸收峰在形成配合物后向低频移动了9~10 cm-1,且吸收强度有一定的变化,同时亚甲基的变形振动吸收峰配位后红移了6~11 cm-1,红外光谱分析进一步证明形成了配合物。
1H NMR:配体和配合物均有芳H、甲基H和亚甲基H 3类核磁共振吸收信号,但配合物与配体相比,芳H的化学位移向低场移动了0.10~0.33,亚甲基上H的化学位移也向低场移动,这说明配体醚氧键上的氧原子参加了配位,使苯环上的电子云密度降低,质子受电子云的屏蔽作用减小,使共振信号向低场方向移动,这与紫外和红外光谱的结论是一致的。
2.3 配合物1的晶体结构
配合物1的晶体结构见图1,晶胞堆积图见图2。配合物1由2个[K(MDB18-C-6)(CH3COCH3)]+配阳离子和1个[Ni(i-mnt)2]2-配阴离子组成,镍原子位于晶体学对称中心,并且不与冠醚中氧原子成键。在[Ni(i-mnt)2]2-配阴离子中i-mnt为二齿配体,2个配体中的4个硫原子与镍原子配位,S(1)-Ni(1)-S(1)i和S(2)i-Ni(1)-S(2)的键角为180°,其他S-Ni-S的键角分别为 78.34(4)°和 101.66(4)°,表明[Ni(i-mnt)2]2-为变形的平面方形构型。 Ni-S、S-C、C≡N、C-C、C=C 间的平均 键 长 分 别 为 0.2209(12)、0.1722(4)、0.1135(6)、0.1479(6)、0.1379(4)nm, 与 配 合 物 [Na(N15-C-5)]2[(Ni(i-mnt)2)]中相应距离基本一致[13]。甲基和苯环上的 C-C间的平均键长为0.148 5(7)nm,与文献[14]中0.149 1(7)nm基本相同。
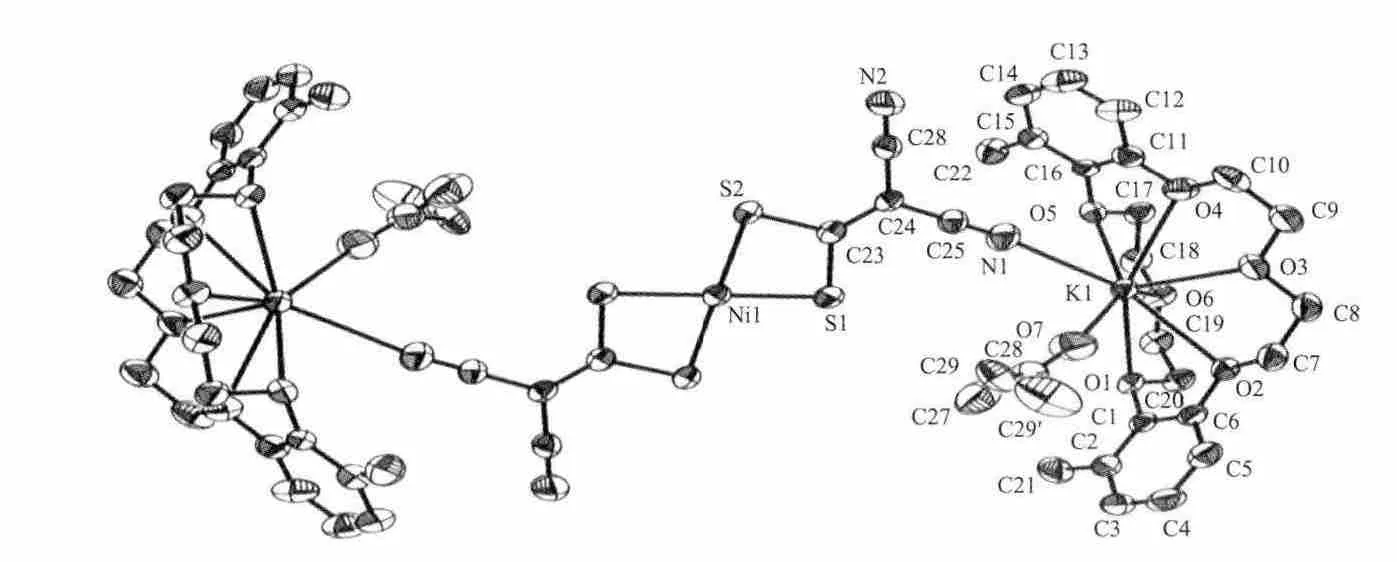
图1 配合物1的分子结构图Fig.1 Molecular structure of the complex 1 shown with 30%probability level displacement ellipsoids

图2 配合物1的晶胞堆积图Fig.2 Packing diagram of the complex 1
在配阳离子[K(MDB18-C-6)(CH3COCH3)]+中,苯环上的2个甲基处于顺式,C(sp2)-O键的平均键长为0.137 9 nm,与文献[13]中0.138 1 nm基本相同,而C(sp3)-O键的平均键长是0.142 0 nm,长于没有取代的二苯并-18-冠-6(0.136 7 nm)[15]。K原子与冠醚中的6个氧原子成键,K-O键的键长在0.265 8(4)~0.287 1(3)nm 范围内,比[K(N18-C-6)]2[(Ni(i-mnt)2]中的相应距离稍短[16],K原子还与丙酮分子中的氧原子和[Ni(i-mnt)2]2-中的N原子成键,K(1)-O(7)键长为0.265 8(4)nm,K(1)-N(1)键长为 0.288 2(4)nm,并且丙酮分子段基C(29)处于无序状态,有C(29)和C(29′)2 个坐标,其中 C(29)占有率为 30.68%,C(29′)的占有率为69.32%。2个[K(MDB18-C-6)(CH3COCH3)]+配阳离子与1个[Ni(i-mnt)2]2-配阴离子形成稳定的中性配合物。
2.4 配合物2的晶体结构
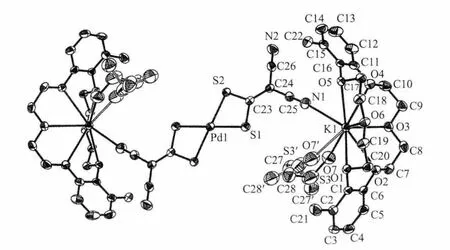
图3 配合物2的分子结构图Fig.3 Molecular structure of the complex 2 shown with 30%probability level displacement ellipsoids
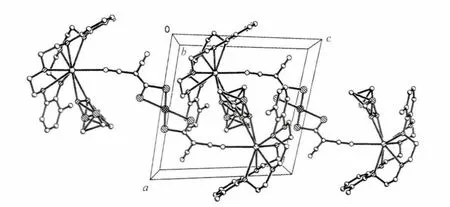
图4 配合物2的晶胞堆积图Fig.4 Packing diagram of the complex 2
配合物2的晶体结构见图3,晶胞堆积图见图4。配合物2也是由2个[K(MDB18-C-6)(CH3SOCH3)]+配阳离子和1个[Pd(i-mnt)2]2-配阴离子组成。配阴离子[Pd(i-mnt)2]2-为变形的平面方形构型。C≡N、C=C、C-C及甲基和苯环上的C-C间的平均键长分别为0.113 3(7)、0.137 9(8)、0.149 3(7)、0.151 0(7)nm,与配合物1中相应距离基本一致。Pd-S间平均键长为0.232 3(13),与[K(DC18-C-6)]2[(Pd(mnt)4)]中相同[17]。 S-C平均键长为0.176 9(6)nm,长于配合物1中的数据,这是因为在配合物2中溶剂二甲亚砜参加了配位。
在配阳离子[K(MDB18-C-6)(CH3SOCH3)]+中,C(sp2)-O和C(sp3)-O键的平均键长分别为0.138 0(6)和0.142 0(5)nm,与配合物1相同。K原子与冠醚中6个氧原子成键,K-O键的键长在 0.267 0(10)~0.291 1(3)nm范围内,比配合物1中的相应距离稍长,但与文献[18]报道的数据基本相同。K原子还与二甲亚砜中的氧原子和[Pd(i-mnt)2]2-中的N原子成键,K(1)-O(7)键长为 0.267 0(10)nm,K(1)-N(1)键长为0.292 2(5)nm,并且与钾配位的二甲亚砜处于无序状态,以S(3)为中心的二甲亚砜分子的空间占有率为69.72%,以S(3′)为中心的二甲亚砜占有率为30.28%。
2.5 配合物热稳定性
配合物1和2的热失重曲线如图5所示。图5表明,2个配合物有相似的热分解过程。它们都是在熔点之前开始分解,失重过程主要有2个阶段:第一阶段,配合物 1在 138.9~211.7℃区间失重为4.85%,相当于失去丙酮分子(理论值为4.43%),配合物2在168.9~236.7℃区间失重为11.78%,相当于失去二甲亚砜(理论值为11.11%)。在第二阶段,配合物1和2分别在232.9℃和317.4℃后,TG曲线出现急剧失重现象,此阶段可能是配合物中冠醚和imnt分子氧化分解所致。随着温度继续升高,配合物1和2至350、400℃以后,TG曲线趋于平坦,说明配合物分解完全,配合物1和2最后稳定在约29.50%和23.83%,残余物为相应配合物的金属氧化物和硫化物,这与Dong等报道的 [K(DC18-C-6)]2[(M(mnt)2)][17]热分解过程基本相同。
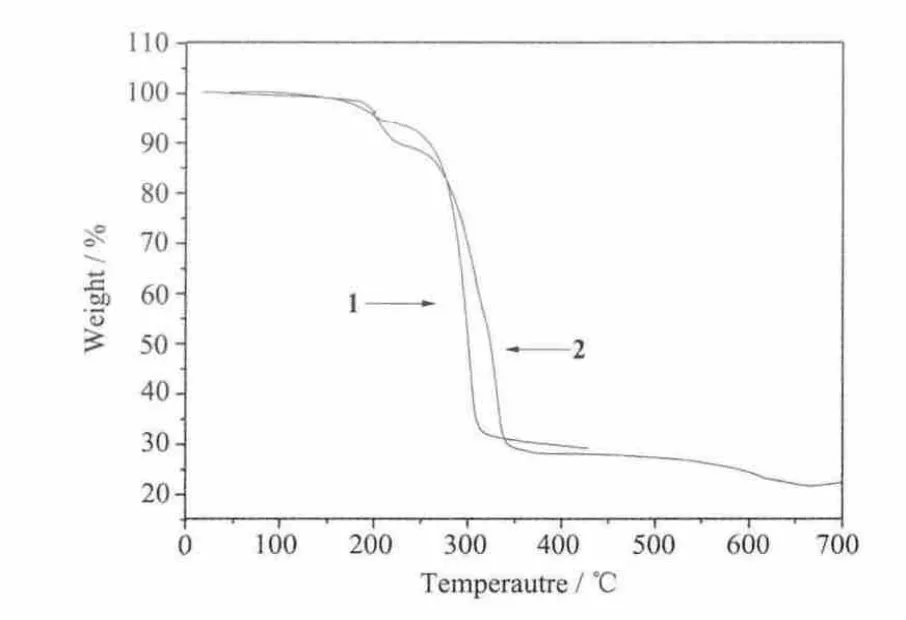
图5 配合物1和2的热失重曲线Fig.5 TG curves of the complex 1 and 2
2.6 配合物与DNA的作用
2.6.1 紫外吸收光谱
文献报道若配合物与DNA发生相互作用,会使配体所处的环境发生变化,从而引起紫外吸收光谱发生改变[19],所以,紫外吸收光谱是研究配合物与DNA相互作用最常用的方法之一。图6为一定浓度的配合物溶液中加入不同浓度的DNA时测定的紫外吸收光谱。由图6可知,随着DNA浓度的增大,紫外吸收光谱发生明显的减色效应,浓度增大,减色效应增强。为了定量研究配合物与DNA的结合强度,由方程 CDNA/(εa-εf)=CDNA/(εb-εf)+1/[Kb(εb-εf)]作图(图 6内嵌),求出配合物和DNA的结合常数Kb[20]。配合物1 和 2 的结合常数分别是:Kb1=4.41×103L·mol-1、Kb2=4.73×103L·mol-1, 远小于以经典插入模式与DNA 结合的 EB(Kb=3.3×105L·mol-1),并且红移效应不太明显,由此可见,配合物可能不是主要以插入模式与DNA作用[20]。
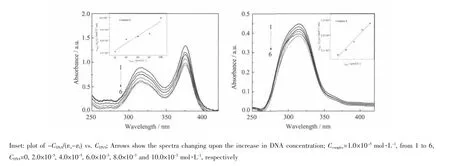
图6 不同浓度CT-DNA存在下配合物1和2的紫外吸收光谱Fig.6 Absorption spectra of the complex in the absence and presence of CT-DNA
2.6.2 荧光猝灭光谱
EB属于芳香族荧光化合物,但本身荧光很弱,当它插入到DNA特定碱基对时,体系的荧光会大大加强。如果配合物能够与DNA作用,就会与EB发生竞争,导致EB-DNA体系的荧光强度减弱,从而产生荧光猝灭。因此,EB可作为配合物与DNA作用的研究探针[21]。图7记录了配合物对EB-DNA体系荧光发射光谱的影响,由图7可知,体系的荧光强度随着配合物浓度的增大而减小,说明配合物取代了EB-DNA体系中一定数量的EB,使该分子从DNA中游离出来,导致EB-DNA体系荧光强度减小。根据 Stern-Volmer方程:I0/I=1+Ksqr[22],式中 I0和 I分别表示未加配合物和加入配合物后体系的荧光强度,r=Ccomplex/CDNA,由 I0/I对 r作图(图 7 内嵌),求得配合物1和2取代EB与DNA作用的荧光猝灭常数分别为:Ksq1=0.56,Ksq2=3.74,说明配合物与DNA发生了相互作用,配合物2的作用能力大于配合物1,这与紫外吸收光谱是一致的,并且与文献[23]报道的Tb(Ⅲ)冠醚配合物Ksq相比要大,说明3-位取代芳香族冠醚过渡金属配合物与DNA作用性能大于稀土冠醚配合物。
2.6.3 圆二色光谱
圆二色光谱(CD)是研究小分子与DNA相互作用的有力工具。DNA的圆二色谱是由不对称的糖分子的螺旋结构产生的,根据配合物对DNA圆二色谱信号的影响特点,不仅可以判断配合物与DNA的相互作用,还可以推断与DNA结合的模式。CT-DNA的CD光谱中有2个明显的特征峰,274 nm左右的正峰是由CT-DNA碱基对π-π堆积作用引起的,245 nm左右的负峰主要对应于双螺旋结构的B型构象[24]。 图8表明了配合物对CT-DNA体系圆二色光谱的影响,由图8可知,加入配合物后,DNA的负峰变化较大,2种配合物都使245 nm的负峰下探增强,并且峰位略有红移,这说明配合物与DNA发生了相互作用,由于274 nm左右的正峰没有明显变化,所以,配合物对DNA碱基对的π-π堆积影响较小[25],配合物不是主要以插入模式与DNA作用,可能是影响了DNA的螺旋结构,导致其构象改变[26],这与紫外和荧光光谱的结论是一致的。

图7 配合物1和2对EB-DNA复合体系荧光光谱的影响Fig.7 Effects of the two complexes on the fluorescence spectra of EB-DNA system
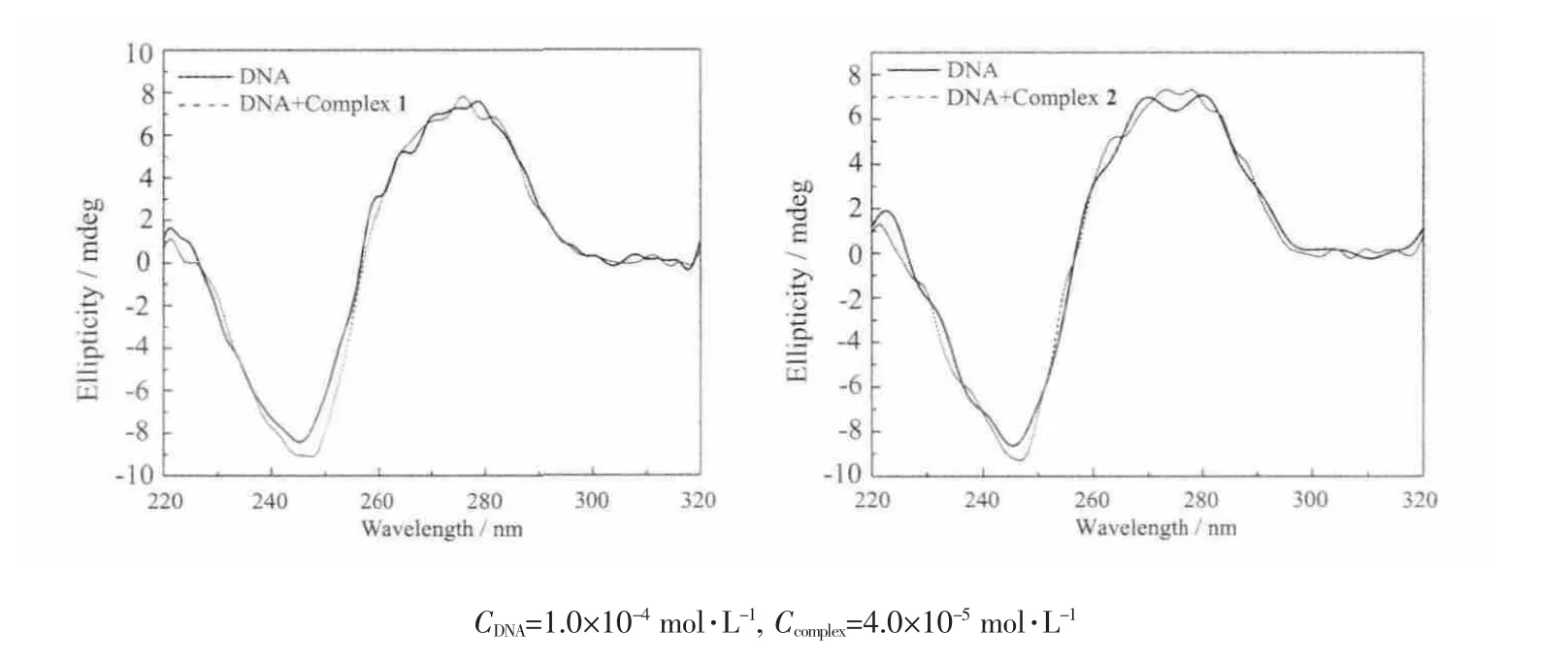
图8 配合物1和2对CT-DNA圆二色光谱的影响Fig.8 Effects of the complexes on CD spectra of CT-DNA
3 结 论
合成了 3,3′-二甲基二苯并-18-冠-6新型冠醚,以合成的冠醚为配体分别与氯化镍、氯化钯和[K2(imnt)]反应,进一步合成了镍(Ⅱ)冠醚配合物1和钯(Ⅱ)冠醚配合物2。通过元素分析、红外光谱、紫外光谱、核磁共振氢谱等测试手段对配体和配合物的结构进行表征。用X-射线单晶衍射测定了配合物的晶体结构。用光谱分析方法初步研究了配合物与CT-DNA的相互作用,结果表明配合物与DNA都具有相互作用,但2种配合物不是主要以插入模式与DNA作用,可能是影响了DNA的螺旋结构,从而引起DNA的构象发生改变,并且配合物2与DNA的作用性能大于配合物1。
[1]Tegoni M X,Remelli M.Coord.Chem.Rev.,2012,256,289-315
[2]Zhang W,Xiong R G.Chem.Rev.,2012,112,1163-1195
[3]MIU Qian(缪 谦),HUANG Hui(黄 辉),HUANG Xiao-Bo(黄小波),et al.Chinese J.Inorg.Chem.(无机化学学报),2009,25(12):2182-2188
[4]Uma H,Shobhana K M.Dyes and Pigm.,2008,77:462-468
[5]LIU Dian-Yi(柳佃义),YAN Xiu-Huan(闫旭焕),TANG Yu(唐瑜),et al.Chinese J.Inorg.Chem.(无机化学学报),2008,24(8):1356-1359
[6]Hasani M,Akbari S.Spectrochim.Acta:Part A,2007,68:409-413
[7]HOU Shou-Jun(侯守君),LI Shu-Mian(李淑勉),TIAN Bao-Zhi(田宝芝),et al.Chinese J.Org.Chem.(有机化学),1992,12:424-428
[8]BAI Yan(白岩),LU Wu-Dang(卢伍党),LI Wen-Guang(李文广),et al.Journal of Lanzhou University:Medical Sciences(兰州大学学报:医学版),2010,36(3):21-25
[9]DONG Li-Bin(董利斌),TIAN Bao-Xin(田宝新),SONG Chong-Li(宋崇立).Chemical Reagents(化学试剂),2000,22(6):362-366
[10]Jensen K A,Henriksen L.Acta Chem.Scand.,1968,22:1107-1128
[11]Sheldrick G M.SHELXTL Version6.10,Bruker Analytical Instrumentation,Madison,Wisconsin,USA,2000.
[12]MENG Ling-Zhi(孟令芝),HE Yong-Bing(何永炳).Organic Spectrum Analyse(有机波谱分析).Wuhan:Wuhan University Press,2002.
[13]Gao X K,Dou J M,Li D C,et al.J.Coord.Chem.,2005,58(17):1597-1605
[14]Niu M J,Wang D Q,Li D C,et al.Z.Kristallogr.-New Cryst.Struct.,2005,220(2):188-190
[15]Bright D,Truter R.J.Chem.Soc.B,1970:1544-1550
[16]Gao X K,Dou J M,Li D C,et al.J.Inclusion Phenom.Macrocyclic Chem.,2005,53:111-119
[17]Dong F Y,Dou J M,Li D C,et al.J.Inorg.Organomet.Polym.Mater.,2005,15(2):119-237
[18]Long E C,Barton J K.Chem.Res.,1990,23:271-277
[19]Strothkamp K G,Strothkamp R E.J.Chem.Educ.,1994,71:77-79
[20]BIAN Lin(边 琳),LI Lian-Zhi(李连之),WANG Xia(王霞),et al.Chinese J.Inorg.Chem.(无机化学学报),2011,27:649-654
[21]GUO Qiong(郭琼),LI Lian-Zhi(李连之),DONG Jian-Fang(董建方),et al.Acta Chim.Sinica(化学学报),2012,70:1617-1624
[22]Lakowicz J R,Weber G.Biochemistry,1973,12:4161-4170
[23]WANG Li-Juan(王丽娟),YANG Tian-Lin(杨天林),YANG Huan-Chun(杨欢春).Chemical Research and Application(化学研究与应用),2010,22(6):683-689
[24]WU Hong-Xing(吴红星),LI Feng-Hua(李风华),LIN Hai(林海),et al.Chinese J.Inorg.Chem.(无机化学学报),2005,21:117-119
[25]Cheng Q R,Pan Z Q,Zhou H,et al.Inorg.Chem.Commun.,2011,14:929-933
[26]LIU Zheng-Jia(刘振佳),SI Yi-Kang(司伊康),CHEN Xiao-Guang(陈晓光),et al.Acta Pharmaceutica Sinica(药学学报),2010,45(12):1478-1484
