合成伏格列波糖的工艺改进
2013-11-19李仲振
李仲振, 张 伟, 王 鹏, 丁 宁, 李 明
(1. 中国海洋大学 医药学院 教育部海洋药物重点实验室,山东 青岛 266003; 2. 复旦大学 药学院,上海 201203)
我国糖尿病(DM)发病率逐年升高[1]。目前,治疗DM的药物主要有几类:α-葡萄糖苷酶抑制剂、磺酰脲类、噻唑烷二酮类、双胍类、餐时血糖调节剂、钒类化合物、胰岛素、调脂治疗药、NO合成酶抑制剂、β3-受体激动剂等。其中,α-葡萄糖苷酶抑制剂[2]作为新一代的降血糖药物,作用机制是通过抑制α-葡萄糖苷酶,减少和推迟肠道吸收碳水化合物,降低餐后高血糖,对Ⅱ型DM进行有效治疗。由于极少的药物吸收,所以肝、肾功能受到的影响较小。已经上市的α-葡萄糖苷酶抑制剂主要包括伏格列波糖(11)、阿卡波糖、米格列醇等。
Takeda公司于1994年在日本推出11(Chart 1)。主要用于抑制餐后高血糖,可以用于预防DM并发症,副作用发生的几率很小[3],是治疗DM一种有效药物。目前11的制备工艺复杂,市场价格昂贵, 因此发展一种11的高效合成方法具有重要意义
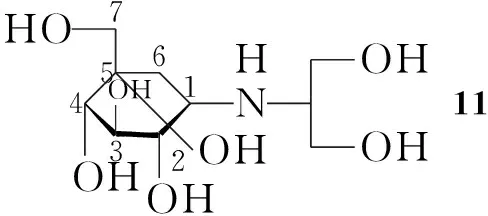
Chart1
合成11主要有两条路线:路线一[4]:以葡萄糖内酯为关键中间体,经还原胺化制备11[5,6];路线二:以6-脱氧葡萄糖-6-烯为原料,经Ferrier重排、亲核加成等反应合成11[7]。路线一成本较低,但制备中间体时操作非常繁琐,需经多次硅胶柱层析纯化,收率较低,致使成本大幅度提高且延长生产周期。
Scheme1
本文对路线一(Scheme 1)进行了工艺改进,简化了后处理方式(如苄基保护以及脱除硫苷的反应),在制备中间体5后,连续合成中间体6~9,而且中间体可以不经柱层析分离纯化直接进行下一步反应,简化了操作,降低了生产成本,使总收率由15.3%[5,6]提高至25.3%。
改进后的路线具有简洁,后处理方便等优点,适合于大规模工业化生产[4]。
1 实验部分
1.1 仪器与试剂
JASCO P-1020型数显旋光仪;JEOL-ECP-600 NMR型核磁共振仪(CDCl3为溶剂,TMS为内标);Q-TOF型质谱仪。
BuLi和Bu3SnCl, Lancaster; CH2Cl2用前经氢化钙回流重蒸;石油醚,60 ℃~90 ℃;薄层色谱硅胶(10 μm~40 μm,显色液8%浓硫酸甲醇溶液),烟台化学工业研究所;柱层析硅胶(300目~400目),青岛海洋化工厂分厂;其余所用试剂均为分析纯。
1.2 合成
(1) 2,3,4,6-四-O-乙酰基-β-D-葡萄糖苯甲硫苷(1)的合成[5]
将葡萄糖一水合物19.8 g(100.0 mmol)和乙酸钠15.0 g(163.0 mmol)混合研磨后,转移至三口瓶中,加入Ac2O 120 mL,搅拌下于120 ℃反应2 h。倒入冰水(300 g)中,剧烈搅拌,有固体析出,置冰浴中静置2 h。抽滤,滤饼用蒸馏水洗涤,用无水乙醇-环己烷重结晶得全乙酰化葡萄糖(A)27.1 g,产率69.5%。
在茄形瓶中加入A 20 g(51.0 mmol)和对甲基苯硫酚9.5 g(76.0 mmol)的CH2Cl2(150 mL)溶液,冰水浴冷却,搅拌下缓慢滴加BF3·Et2O 13 mL,滴毕,于室温反应5 h(TLC检测)。加入蒸馏水(50 mL)稀释,慢慢加入固体NaHCO3至pH 7。用CH2Cl2(500 mL)稀释后,分别用饱和NaHCO3溶液(2×50 mL),饱和NaCl溶液(2×50 mL)洗涤,无水Na2SO4干燥;减压浓缩,残余物用无水乙醇-环己烷重结晶得白色针状晶体1 21.4 g,收率92.3%;1H NMRδ: 7.22~7.33(m, 4H, PhH), 5.21(t,J=9.5 Hz, 1H, 3-H), 5.02(t,J=9.9 Hz, 1H, 4-H), 4.94[t(apparent splitting pattern),J=9.5 Hz, 9.9 Hz, 1H, 2-H], 4.64(d,J=10.3 Hz, 1H, 1-H), 4.17~4.23(m, 2H, 6-H), 3.71~3.69(m, 1H, 5-H), 2.40(s, 3H, PhCH3), 2.01, 2.01, 2.10(s, 12H, CH3in Ac)。
(2)β-D-葡萄糖苯甲硫苷(2)的合成[4]
在茄形瓶中加入1 17.6 g(40.0 mmol)的MeOH/CH2Cl2(V/V=1/1, 200 mL)溶液,搅拌下加入MeONa 50 mg,于室温反应30 min(TLC检测)。加入阳离子交换树脂中和,过滤,滤液减压浓缩得淡黄色糖浆(2),直接进行下步反应。
(3) 2,3,4,6-四-O-苄基-β-D-葡萄糖苯甲硫苷(3)的合成[4]
将2溶于DMF(200 mL)中,冰水浴冷却,搅拌下分批加入NaH 10.4 g(6.5 eq),加毕,于室温反应30 min。冰水浴冷却,滴加BnBr 28.5 mL(6.0 eq),滴毕,反应1 h;于室温反应反应6 h(TLC检测)。冰水浴冷却,缓慢加入蒸馏水(50 mL)终止反应(出现大量气泡)。用1 mol·L-1盐酸调至pH<1,出现大量黄色颗粒,置冰箱(-18 ℃)中静置3 h。过滤,滤饼用冷水洗涤,干燥得黄色固体3 25.2 g,收率100%(两步);1H NMRδ: 7.01~7.49(m, 24H, PhH), 4.54~4.90(m, 8H, PhCH2), 4.73(d,J=10.1 Hz, 1H, 1-H), 3.78(dd,J=10.1 Hz, 9.2 Hz, 1H, 2-H), 3.67~3.73(m, 1H, 5-H), 3.69(t,J=9.2 Hz, 1H, 4-H), 3.63(t,J=9.2 Hz, 1H, 3-H), 3.46~3.49(m, 2H, 6-H), 2.30(s, 3H, PhCH3)。
(4) 2,3,4,6-四-O-苄基-葡萄糖(4)的合成[4]
在反应瓶中加入3 19.4 g(30.0 mmol)的丙酮/水(V/V=5/1, 500 mL)溶液,搅拌下分批加入N-溴代丁二酰亚胺(NBS)18.7 g(105.0 mmol),加毕,于室温反应30 min(TLC检测)。减压蒸除大部分丙酮,析出白色固体,加入1 mol·L-1盐酸(200 mL)稀释,置冰箱(-18 ℃)中静置3 h。抽滤,滤饼用冷无水乙醇洗涤,用无水乙醇-环己烷重结晶得白色絮状固体4 15.3 g,收率95%。
(5) 2,3,4,6-四-O-苄基-葡萄糖内酯(5)的合成[4]
在反应瓶中加入4 5.4 g(10.0 mmol)的DMSO(40 mL)溶液,搅拌下滴加Ac2O 20 mL,滴毕,于室温反应12 h。加入饱和NaHCO3中和Ac2O;加入CH2Cl2100 mL,依次用水(2×25 mL)和饱和NaCl溶液(2×25 mL)洗涤,无水Na2SO4干燥,减压浓缩得黄色油状液体5 5.12 g,收率95.1%;1H NMR(DMSO-d6)δ: 7.49~7.01(m, 20H, PhH), 4.87~4.48(m, 8H, PhCH2), 4.60(td,J=7.8 Hz, 3.2 Hz, 1H, 5-H), 4.36(d,J=6.5 Hz, 1H, 2-H), 4.02(t,J=6.4 Hz, 1H, 3-H), 3.88(dd,J=7.8 Hz, 6.5 Hz, 1H, 4-H), 3.68(dd,J=11.0 Hz, 3.1 Hz, 2H, 6-H)。
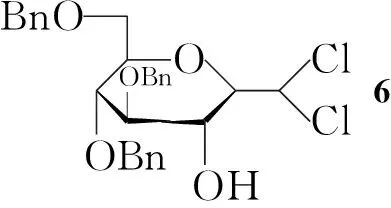
(6) (2R,3R,4S,5R,6R)-3,4,5-三-苄基-6-苄氧甲基-2-二氯甲基-四氢-2H-吡喃-2-醇(6)的合成[4]
在反应瓶中加入二异丙基胺850μL(6.0 mmol)的干燥THF(6 mL)溶液,冰盐浴冷却至-5 ℃~-10 ℃,搅拌下滴加丁基锂的正己烷溶液2.4 mL(6.0 mmol),滴毕,反应1 h得LDA溶液。
在反应瓶中加入5 1.60 g(3.0 mmol)的CH2Cl2(15 mL)溶液,冷却至-70 ℃,搅拌下滴加LDA溶液,滴毕,于-70 ℃反应1 h(TLC检测)。加入1 mol·L-1盐酸20 mL终止反应,用CH2Cl2(200 mL)稀释,依次用1 mol·L-1HCl(2×30 mL),饱和NaHCO3溶液(2×30 mL),饱和食盐水(2×20 mL)洗涤,无水硫酸钠干燥,减压浓缩得淡黄色油状液体,经硅胶柱层析[梯度洗脱剂:A=V(石油醚) ∶V(乙酸乙酯)=10 ∶1~6 ∶1)]纯化得淡黄色油状液体6 1.84 g,收率99.5%;1H NMRδ: 7.22~7.38(m, 20H, PhH), 5.81(s, 1H, CHCl2), 4.60~4.97(s, 8H, PhCH2), 4.06(t,J=9.2 Hz, 1H, 3-H), 3.98~4.01(m, 2H, 2,5-H), 3.83(dd,J=11.7 Hz, 4.0 Hz, 1H, 6-Ha), 3.76(t,J=9.3 Hz, 1H, 4-H), 3.72(dd,J=11.7 Hz, 1.9 Hz, 1H, 6-Hb), 3.33(s, 1H, OH)。
(7) (3R,4S,5R)-3,4,5,7-四-苄氧基-1,1-二氯庚烷-2,6-二醇(7)的合成[4]
在反应瓶中加入61.84 g(2.9 mmol)的二甲醚(DME, 20 mL)溶液,冰浴冷却下分批加入NaBH4190 mg(5.0 mmol),加毕,于室温反应6 h。减压浓缩,残余物加水(20 mL)分散,用乙酸乙酯(2×50 mL)萃取,合并萃取液,依次用1 mol·L-1盐酸(2×15 mL),饱和NaHCO3溶液(2×15 mL)洗涤,用无水Na2SO4干燥后浓缩得淡黄色油状液体7 1.69 g,收率92.0%。7存在两种异构体,无需分离,直接进行下一步反应。
(8) (2R,3S,4S,5S)-4,5,6-三-苄氧基-3-苄氧甲基-2,2-二-氯-3-羟基环己酮(8)的合成[4]
在反应瓶中加入DMSO 1.2 mL(8 eq)和CH2Cl215 mL,搅拌使其混合;冷却至-70 ℃,滴加三氟乙酸酐(TFAA)1.16 mL(4 eq)的CH2Cl2(10 mL)溶液,滴毕,反应30 min;滴加7 1.18 g(1.9 mmol)的CH2Cl2(10 mL)溶液,滴毕,反应1 h。加入Et3N 2.3 mL,反应15 min。逐渐升温至0 ℃,加入冰水(10 mL),搅拌15 min后加入CH2Cl2(100 mL)稀释,分别用1 mol·L-1盐酸(2×15 mL)、饱和NaHCO3溶液(2×20 mL)、饱和食盐水(2×20 mL)洗涤,经无水Na2SO4干燥后浓缩,残余物经柱层析(梯度洗脱剂:A=12 ∶1~6 ∶1)纯化得白色固体8 958 mg,收率74.2%;1H NMRδ: 7.42~7.22(s, 20H, PhH), 4.98~4.55(s, 8H, PhCH2), 4.92(d,J=9.5 Hz, 1H, 4-H), 4.29(t,J=9.2 Hz, 1H, 2-H), 4.07(t,J=9.5 Hz, 1H, 3-H), 3.84(m, 2H, 7-H), 3.37(s, 1H, OH)。
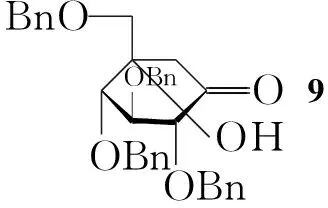
(9) (2R,3S,4S,5S)-2,3,4-三-苄氧基-5-苄氧甲基-5-羟基环己酮(9)的合成[4]
在反应瓶中加入三丁基氯化锡1.50 g的乙醚(5 mL)溶液,冰水浴冷却,搅拌下分批加入LiAlH460 mg,加毕,反应15 min;升至室温,反应3 h。加适量冰水,搅拌至无明显气泡冒出为止。加入乙醚50 mL,用冰水(2×25 mL)洗涤,无水Na2SO4干燥后浓缩得无色澄清透明液体三丁基锡,Ar气氛下保存备用。
在反应瓶中加入8 745.2 mg(1.2 mmol)的干燥甲苯(10 mL)溶液,搅拌下加入偶氮二异丁腈(AIBN)40 mg(0.24 mmol),三丁基锡1.22 g(4.2 mmol),氩气保护下升温至100 ℃反应1 h。冷却至室温,加入乙酸乙酯50 mL,分别用1 mol·L-1盐酸(2×20 mL)、饱和NaHCO3溶液(2×20 mL)洗涤,无水Na2SO4干燥,减压浓缩,残余物经柱层析(梯度洗脱剂:A=8 ∶1~3 ∶1)纯化得白色固体9 494.8 mg,收率74.6%;1H NMRδ: 7.40~7.26(s, 20H, PhH), 5.00~4.40(s, 8H, PhCH2), 4.14(d,J=9.5 Hz, 1H, 4-H), 4.06(d,J=9.2 Hz, 1H, 2-H), 4.01(t,J=9.2 Hz, 1H, 3-H), 3.53(d,J=8.5 Hz, 1H, 7-Hb), 3.16(d,J=8.5 Hz, 1H, 7-Ha), 2.83(d,J=14.6 Hz, 1H, 6-Hb), 2.48(d,J=14.7 Hz, 1H, 6-Ha), 2.39(d,J=2.2 Hz, 1H, OH)。
(10) (1S)-1-(羟基)-2,3,4-三-苄氧基-5-[2-羟基-1-(羟甲基)乙基氨基]-1-碳-苄氧基甲基-1,2,3,4-环己烷四醇(10)的合成[4]
(11) 11的合成[4]
2 结果与讨论
以葡萄糖内酯为关键中间体进行11合成的工艺路线主要包括三个主要部分(Scheme 1)[4],合成葡萄糖内酯(5)、制备环己酮(8)及胺化还原得11。
在合成5时,首先必须获得异头位羟基裸露的化合物4。通过查询文献,我们发现葡萄糖异头位常用的保护基有OMe, allyl(OCH2CH=CH2), propargyl(OCH2C≡C)以及SR(R=Me, Et or Ph)等。采用OMe保护异头位,在脱除OMe时收率较低[8~10],且部分Bn同时被脱除,分离纯化非常困难,操作较为复杂;采用OCH2CH=CH2, OCH2C≡C保护基时,脱除时收率较高,但需要使用昂贵的催化剂如Pd(PPh3)4[11]及AuCl3[12]等,导致成本高昂,同时催化剂难以回收利用,不利于工业化生产;异头位的硫苷保护基在脱除时可以采用廉价易得的NBS或NIS,收率较高,是一种理想的保护基。但由于采用SEt或者SPh保护异头碳的中间体多为油状液体或糖浆[13],难以固化,不能用重结晶方法纯化,需采用柱层析分离,并且会出现令人不适的味道,污染环境。
针对上述问题,本文采用对甲苯硫苷保护葡萄糖的异头位,不仅可以避免不愉快的味道,且产物为固体,可通过重结晶方法纯化,极大地提高了效率。全乙酰化葡萄糖在BF3·Et2O催化下与对甲苯硫酚发生糖苷化反应后,用无水乙醇-环己烷重结晶,以92.3%的高收率得到1。接着用MeONa脱除1中的乙酰基保护基得到2; 2后经苄基保护、脱硫苷得到异头位羟基裸露的苄基保护的4。
在氧化条件下,4的羟基氧化为羰基制备5。常用氧化体系为Dess-Martin氧化剂,二氯甲烷为溶剂,25 ℃反应,收率89%[14]; 4-甲基-N-氧化物/四丙基高钌酸铵体系,二氯甲烷为溶剂,加入分子筛作为吸水剂, 20 ℃反应,收率86%[15]。本文采用廉价易得的DMSO-Ac2O体系可以使收率提高到95%以上,并且反应条件非常温和,对空气及水汽不敏感,四步总收率83.0%(以1计)[16]。所有操作简便易行、无需柱层析纯化。
对5的羰基进行加成反应制备6的反应必须严格防水。适当延长反应时间(由60 min延长到90 min),收率可以由文献90%[5]提高到近乎定量。半缩酮6经NaBH4还原为直链醇,得到的开环产物7为差向异构体混合物,可以不经分离直接用于下一步反应。
合成8时[5],首先采用Swern氧化(DMSO/TFAA体系)将两个游离羟基氧化为酮,之后加入有机碱Et3N发生分子内的羟醛缩合反应形成8,收率74.2%(与文献收率相当)。
利用Bu3SnH在引发剂AIBN作用下自由基历程将8分子中的氯还原制备环己酮9。 9与2-氨基-1,3-丙二醇首先反应制得席夫碱后经还原剂还原后可得到10。使用传统的还原方法(NaBH4),以约70%的收率得到α-和β-两种异构体的混合物,分离纯化比较困难。实验中我们发现,改用NaBH3CN做还原剂时,以69%的收率、高立体选择性得到所需的唯一α-构型产物。这一改进避免了复杂的分离过程,极大的提高了生产效率。
通过合适的方法去除10中的苄基保护基就可以得到11。文献[6]方法采用HCO2H作为氢供体,Pd-black作为催化剂室温下反应得到11。该方法的缺点是Pd-black价格高昂,且用量很大,约为底物质量的20%。本文尝试对其进行了改进,换用廉价的10%Pd-C作为催化剂,通过比较甲醇-甲酸、EtOAc溶剂体系,我们发现使用体积比为15 ∶1的甲醇-甲酸做为溶剂效果最好。该溶剂体系对原料和产物均有较好的溶解能力,可以缩短反应时间、提高反应收率。后处理过程中,文献需要通过反复柱层析(先后经阳离子交换树脂柱及阴离子交换树脂柱纯化)才能够得到目标纯品,操作繁琐复杂;而我们在经过中和甲酸、过滤及DCM洗涤后即可达到纯度要求,极大的提高了效率,降低了生成成本[4]。
[1] Wenying Yang, Juming Lu, Jianping Weng,etal. Prevalence of diabetes among men and women in china[J].N Engl J Med,2010,(362):1090-1101.
[2] 谌卫. 降血糖药物及其作用靶点研究[J].中国误诊学杂志,2011,11:8845-8847.
[3] Andrade R J, Lucena M, Vega J L,etal. Acarbose-associated hepatotoxicity[J].Diabetes Care,1998,21:2029-2030.
[4] 李仲振. 地高辛皂苷和岩藻寡糖的合成研究[D].青岛:中国海洋大学,2009.
[5] Hiroshi F, Satoshi H. Synthesis of a branched-chain inosose derivative,a versatile synthon ofN-substituted valiolamine derivatives from D-glucose[J].J Org Chem,1992,57:3642-3650.
[6] Hiroshi F, Satoshi H. Synthesis of valiolamine and itsN-substituted derivatives AO-128,validoxylamine G,and validamycin G via branched-chain inosose derivatives[J].J Org Chem,1992,57:3651-3658.
[7] Boyer F D, Lallemand JY. Enantioselective syntheses of polyhydroxylated nortropane derivatives:Total synthesis of (+) and (-)-calystegine B2[J].Tetrahedron,1994,50(35):10443-10458.
[8] Mandai T, Okumoto H, Oshitari T,etal. Synthesis and biological evaluation of water soluble taxoids bearing sugar moieties[J].Heterocycles,2001,54:561-566.
[9] Bernardes G J L, Thompson S, Chalker J M,etal.From sisulfide- to thioether-linked glycoproteins[J].Angew Chemie Int Ed,2008,47(12):2244-2247.
[10] Mark M, Staveski B Y F, Wan Alan E. Walts,phospholipid-saccharide conjugates,genzyme corporation[P].US 5 354 853,1994.
[11] Tsukamoto H, Kondo Y. Facile and selective cleavage of allyl ethers based on palladium(0)-catalyzed allylic alkylation ofN,N′-dimethylbarbituric acid[J].Synlett,2003,7:1061-1063.
[12] Hotha S, Kashyap S. Propargyl glycosides as stable glycosyl donors:Anomeric activation and glycoside syntheses[J].J Am Chem Soc,2006,128:9620-9621.
[13] Dinkelaar J, Witte M D, Bos L J,etal. NIS/TFA:A general method for hydrolyzing thioglycosides[J].Carbohy Res,2006,(341):1723-1729.
[14] René Csuk, Petra Dörr. Convenient oxidations of carbohydrate derived lactols and ofε-hydroxy-β-ketophospnates[J].J Carbohydr Chem,1995,14:35-44.
[15] Lee J, Lee S, Seo H J,etal. NovelC-aryl glucoside SGLT2 inhibitors as potential antidiabetic agents:1,3,4-Thiadiazolylmethylphenyl glucoside congeners[J].Bio Med Chem,2010,18(6):2178-2194.
[16] Hiroyoshi K, Hewitt G, Fletcher J. Syntheses with partially benzylated sugars.Ⅷ.Substitution at carbon-5 in aldose.The synthesis of 5-O-methyl-D-glucofuranose derivatives[J].J Org Chem,1967,32(8):2531-2534.
