一个RNA剪接调控元件分类方法的研究
2012-11-27王泽锋
马 猛 汪 洋 汝 颖 王泽锋*
1(北卡罗来纳大学教堂山分校药理学系,教堂山27599-7365,美国)
2(安徽大学计算机科学技术学院,合肥 230039)
3(安徽省立医院内分泌科,合肥 230001)
引言
RNA剪接调控元件在RNA剪接过程中起着重要的调控作用,正确识别RNA剪接调控元件对深刻理解RNA剪接过程、认识人基因组生物复杂性具有重要意义。目前已有数种不同的计算[1-3]和生物[4]方法可用来识别外显子中的RNA剪接元件,这些方法虽具有较低的假阳性率,但其假阴性率难以确定,所以不排除还有部分剪接元件未被识别,如何发现这些未被识别的剪接元件是个有趣的问题,序列分类方法可被用于分析解决该问题。
目前已有多种分类方法被用于各种生物序列数据的分析:决策树[5-6]、神经网络[7]、支持向量机[8-9]、朴素贝叶斯[10]、隐马尔可夫模型[11]、k-近邻法[12]、PSSM[13]等。本研究设计了一个新的基于序列特征的分类方法,并将该方法用于剪接调控元件的分析。外显子剪接增强子(ESE)和沉默子(ESS)是两类重要的基因剪接调控元件,针对已知ESE和ESS八联体,该方法首先从已知剪接元件中抽取序列特征,查找未知元件满足的序列特征,计算未知元件的剪接分值,最后对未知元件的剪接功能进行预测,该方法不仅可以给出预测结果,还可以给出预测依据,即元件中包含的序列特征以及共享这些序列特征的已知剪接元件,透明的预测结构利于进行生物解释。该方法也可以被用于分析其他类型的生物序列数据,如蛋白质序列数据等。
1 数据准备
将基于八联体剪接调控元件构建分类器。数据来源于文献[2],其中包含2060个 ESE、1016个ESS,每个剪接元件都是一个含有八个核苷酸的顺序序列,如表1所示的 ESE八联体数据,每行分别表示从第一位到第八位的核苷酸。
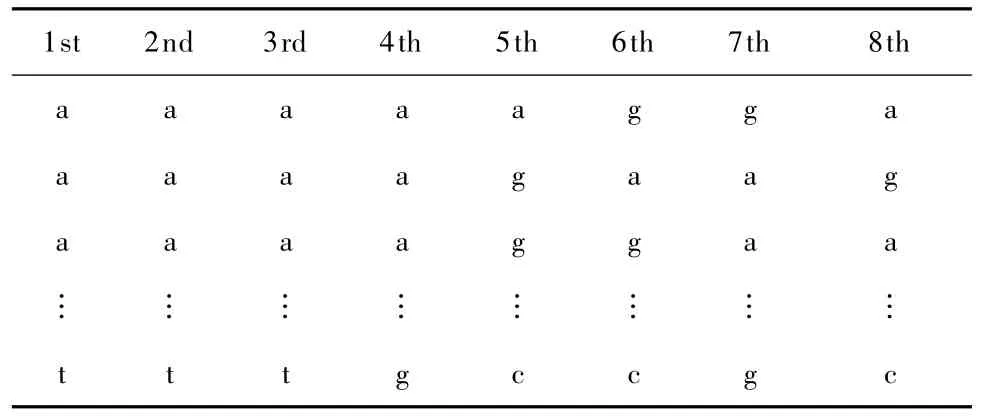
表1 ESE八联体数据Tab.1 Trusted ESE 8mers
2 抽取序列特征
具有相似剪接功能且为同类剪接因子识别的RNA剪接元件往往表现出类似的序列特征,而具有不同剪接功能的剪接元件则表现出较大差异的序列特征,这提示剪接元件中的序列特征含有丰富的分类信息,可用来分类剪接元件、预测未知元件的剪接功能。
下面将以ESE八联体数据为例介绍如何从剪接元件中抽取序列特征。为了便于形式化描述,参考关联规则挖掘[14-15],给出剪接元件序列特征挖掘的相关定义。
定义1.项目集I
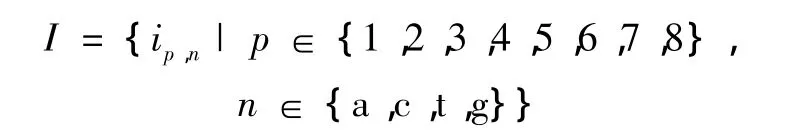
项目ip,n表示在八联体的第 p个位置出现了核苷酸n。项目集I共包含32个项目。
定义2.包含k个项目的I的非空子集称为 k项集。任何一个八联体o都是满足如下约束的八项集:
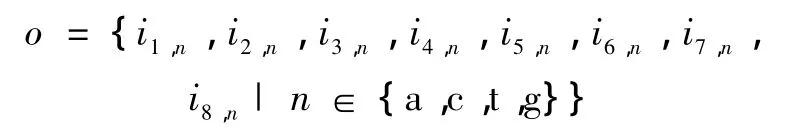
例如表示 ESE八联体 aaaaagga的 8项集是{i1,a,i2,a,i3,a,i4,a,i5,g,i6,a,i7,g,i8,a},可简写为 i1ai2ai3ai4ai5gi6ai7gi8a。
定义3.对 ESE八联体数据集 DESE和项集 is,DESE中包含项集 is的八联体数目记为 ris,则定义 is在DESE中支持度如下:
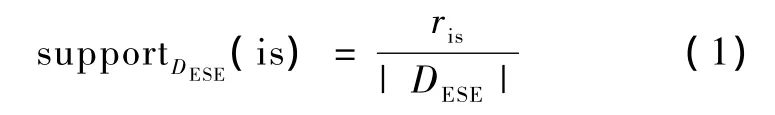
式中,|DESE|表示数据集 DESE中包含的八联体个数。
定义4.对DESE和项集is,给定最小支持度阈值minsup,如果supportDESE(is)≥minsup,则称is为频繁项集,在文中也将其称为序列特征,如果 is中包含k个项目,则称is为频繁k项集或k项序列特征。这些序列特征可看作是ESE或ESS的保守模式,当一个元件中包含这些序列特征时,则它具有较大概率可以表现出ESE或ESS功能,但有时,即使包含这些保守模式,元件也没有表现出某种剪接调控功能,为了解释这个问题,引入扩展序列特征。
3 扩展序列特征
不同类别RNA剪接元件具有不同的序列特征,序列特征支持度的大小可理解为该序列特征对剪接功能类别标记作用的强弱。例如ESE序列特征i1ti2ti3t的支持度为 12.01%,而 i1ai2ai3a的支持度为5.02%,则称i1ti2ti3t对ESE剪接类别的标记作用强于i1ai2ai3a。
序列特征指出了在八联体中的某些特定位置上出现的核苷酸,这些特定位置称为该序列特征的保守位置,其余的称为灵活位置,那么灵活位置上核苷酸的出现频率是否有差异呢?下面我们以ESE序列特征i1ti2ti3t为例,从 DESE中统计该序列特征的灵活位置(4、5、6、7、8)上 4 种核苷酸的出现频率结果如表2所示。
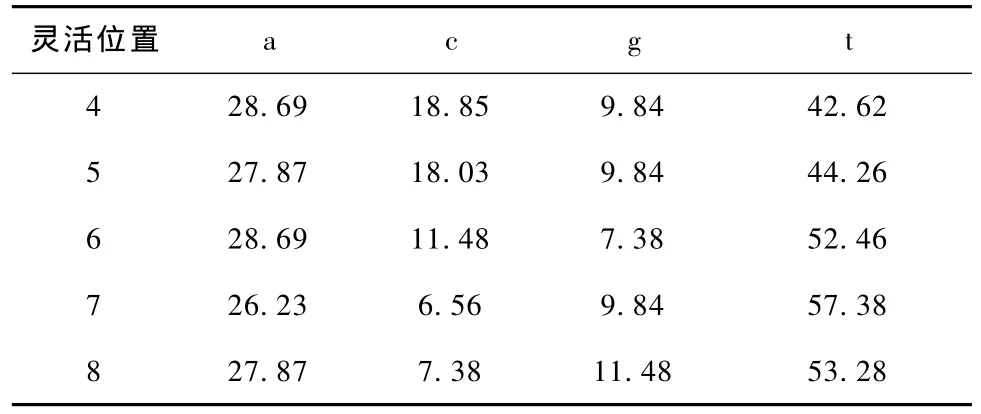
表2 在 i1ti2ti3t的灵活位置(4,5,6,7,8)上核苷酸的出现频率(%)Tab.2 The nucleotide frequencies(%)in the flexible positions of i1ti2ti3t
如果灵活位置上的核苷酸与元件的剪接调控功能无关,则该位置上的核苷酸分布应该是随机的,即每种核苷酸出现频率期望值应该为25%,但从表2中可以看到,4种核苷酸在i1ti2ti3t的灵活位置上的分布表现出明显差异,例如在位置7,核苷酸 t的频率为57.38%,而c的频率只有6.56%,这提示序列特征灵活位置上核苷酸的分布也包含有丰富的分类信息可以利用。所以对一个八联体,不仅要考察它是否满足序列特征,还要考察它在灵活位置上出现的核苷酸以及相应的概率。
基于以上观察,下面将给出扩展序列特征的相关定义。
定义5.ESE序列特征sf包含m个保守位置、k个灵活位置,可采用类似正则表达式的形式,将该序列特征灵活位置上的核苷酸分布信息与该序列特征连接,得到扩展序列特征esf,例如ESE序列特征i1ti2ti3t的扩展序列特征为:
i1ti2ti3t[a:28.69、c:18.85、g:9.84、t:42.62][a:27.87、c:18.03、g:9.84、t:44.26][a:28.69、c:11.48、g:7.38、t:52.46][a:26.23、c:6.56、g:9.84、t:57.38][a:27.87、c:7.38、g:11.48、t:53.28]
定义6.定义扩展序列特征esf与序列特征sf具有相同支持度,即:
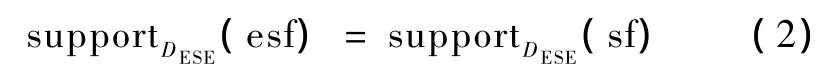
如果某八联体o包含序列特征sf的所有项目,则称o满足序列特征sf及其扩展esf。
定义7.给定八联体o及包含m个保守位置k个灵活位置的 ESE扩展序列特征 esf,如果 o满足esf,则o满足esf的概率计算如下:
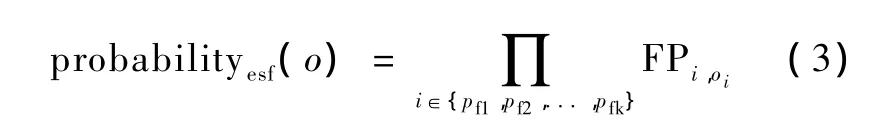
其中,{pf1,pf2,…,pfk}为 esf的灵活位置集合,esf灵活位置上核苷酸出现频率保存在数列FP中,oi表示八联体o第 i个位置上出现的核苷酸,FPi,oi表示核苷酸oi在灵活位置i上的出现频率。
在得到ESE和ESS的扩展序列特征之后,可将未知元件与不同类别的扩展序列特征进行比较以来推断该元件的剪接功能。如果一个元件满足某个剪接功能类别的特征,则可解释为该元件具有表现出此类剪接功能的潜在能力,但实验中,发现很多元件同时满足多个不同剪接功能类别的特征,此时该如何判断该元件的剪接功能呢?解决该问题的基本想法是判断在该元件满足的特征中,哪种类别的特征更强,然后依此推断该元件更倾向表现出的剪接功能。基于该思想,下一节引入剪接分值的概念,由此来推断未知元件的剪接功能。
4 计算剪接分值
本节中,将基于元件中蕴含的扩展序列特征,计算元件的剪接分值,推测其剪接功能强弱。
定义8.给定ESE八联体数据集DESE,八联体o所满足的全部ESE扩展序列特征记为ESEF(o),则o的ESE剪接分值计算如下:
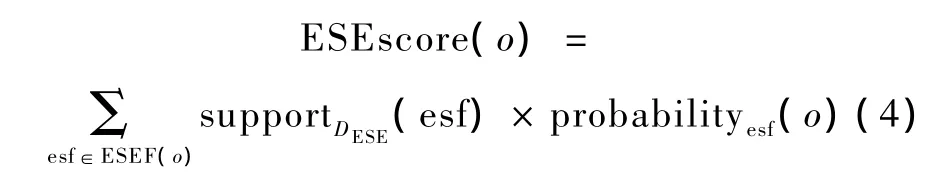
ESE剪接分值的计算考虑到了八联体o满足的序列特征数、序列特征的支持度和序列特征灵活位置上核苷酸分布的差异。八联体 o的ESE剪接分值越高,则表现出ESE剪接功能的概率越大。类似的方法可计算出ESS剪接分值。
下一节将介绍倾向指数的概念用于预测未知元件的剪接功能。
5 预测未知元件的剪接功能
目前已有多种计算或生物的方法被用来识别RNA剪接元件,这些方法虽然都具有较低的假阳性率,但不排除还有剪接元件未被发现,本研究利用剪接分值将ESE和ESS分成两簇,而对于那些落于ESE或ESS簇内的未知八联体,说明该元件包含ESE或ESS的序列特征,有理由怀疑它们具有某种程度的ESE或者ESS的剪接功能。本节将介绍如何基于剪接分值预测未知八联体的剪接功能
首先定义倾向指数。对八联体 o,计算得到ESE和ESS剪接分值,则o的倾向指数为
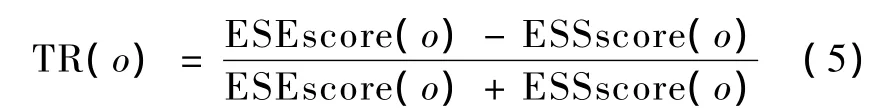
o倾向指数 TR的变化范围是[-1,+1],如果TR是个正小数,则提示八联体 o倾向表现为 ESE剪接功能,如果TR是个负数,则提示八联体o倾向表现为ESS剪接功能。
6 实验
采用的2060个ESE和1016个ESS八联体实验数据来自文献[2]。ESE和ESS中的序列特征挖掘类似关联规则分析中的频繁项集挖掘。Agrawal等于1993年提出关联规则挖掘问题[15],并设计出Apriori算法以解决该问题[14]。产生频繁项集是Apriori算法的核心步骤。将基于 Apriori算法从剪接元件数据中挖掘出全部序列特征。定义最小支持度阈值为5%,对ESE八联体数据集而言,任何支持度计数大于103(2060×0.05)的项集都认为是ESE序列特征,最后挖掘得2375个ESE序列特征。将该方法应用于 ESS八联体数据集,得到1515个ESS序列特征。由于不同长度的序列特征是重叠的,例如i1ti2ti3t与 i1ti2t重叠,所以,仅基于长度为 2的序列特征计算已知元件的ESE和ESS剪接分值。如果某八联体剪接分值小于10-7,则定义其剪接分值为10-7。图1是对数刻度下已知八联体的 ESE和ESS剪接分值分布图。如图1所示,98%的可信ESE八联体的ESE剪接分值大于10-7,94.5%的可信ESS八联体的ESS剪接分值大于10-7,从图1剪接分值的分布可以看到,ESE和 ESS被清晰分开,剪接分值显示出了良好的剪接元件分类能力。
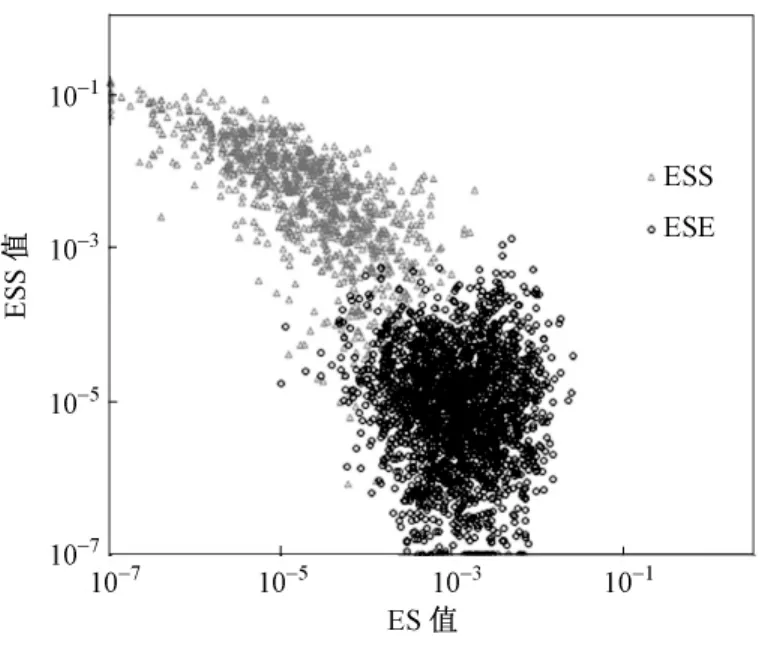
图1 已知八联体ESE和ESS剪接分值分布Fig.1 Splicing score distribution for known ESE and ESS octamers
计算所有ESE和ESS的TR指数,并分区统计分布,结果见图2。从图2可以看出,95.5%的 ESE的TR指数大于0.6,85.5%的 ESS的TR指数小于-0.6,这表明TR指数对ESE和ESS有明确的标记功能。根据以上结果,可以构建一个简单的分类器如下:基于可信的ESE和ESS八联体,对任一未知八联体o,计算其TR指数。这里采取一个较宽松的阈值0.5,如果 TR>0.5,则预测 o倾向表现为 ESE剪接功能;如果 TR<-0.5,则预测 o倾向表现为ESS剪接功能;否则,难以断定o的剪接功能。为了验证该分类器的有效性,获得对分类错误率的可靠估计,采用了3种方法进行验证实验。
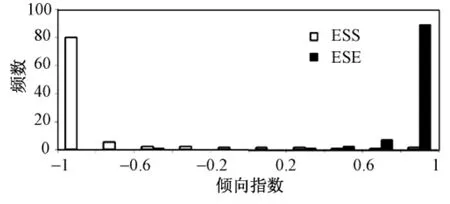
图2 ESE和ESS的TR指数统计直方图Fig.2 Histogram ofTR forESE and ESS octamers
1)将ESE和ESS八联体数据集混合,在混合数据集上采用留一法进行样本类别预测,即从数据集上每次保留一个不同的八联体作为测试样本,其余作为训练样本,然后基于训练样本构建分类器,对测试样本进行类别预测,重复该过程,直到所有ESE和ESS都被作为一次测试样本为止。
2)从ESE和ESS数据集中分别随机抽取20%的数据作为测试集,有615个样本,其余作为训练集,基于训练集构建分类器,对测试集中的每个样本进行类别预测,该过程称为独立测试实验。
3)将混合数据集分成10个子集,其中的9个子集,每个子集有307个八联体,包含206个 ESE和101个ESS,第10个子集包含206个 ESE和107个ESS。选择一个子集作为测试集,其余作为训练集,重复该过程,直到所有的子集均被用作一次测试集。这个过程称为分层十折交叉验证实验。每个子集上错误分类数之和为实验总的错误分类数。
还采取了另外3种方法对ESE和ESS的实验数据进行了上述验证实验:k-近邻[16]、决策树[17]和朴素贝叶斯方法[18],实验结果见表3。从实验结果来看,k-近邻方法的预测精度最高,在留一法、独立测试和分层十折交叉验证实验中,预测精度均高于97%,其次是本方法,3个验证实验的预测精度都在93%左右,朴素贝叶斯方法预测精度最低。这3个验证实验说明本方法具有较好的预测精度和健壮性。从基于元件距离进行预测的角度来看,本方法与k-近邻法具有相似的预测思想,即相似者具有相似功能。这里k-近邻方法采用的是海明距离来度量元件间的距离,而本方法采用的是序列特征,将元件中包含的不同功能的序列特征的多少和强弱量化为剪接分值,来预测元件功能。利用本方法对未知元件的剪接功能进行预测时,不仅可以给出预测结果,还可以给出预测的依据,即该元件中包含的序列特征和共享该特征的已知剪接元件,这有助于对未知元件的剪接功能进行生物解释。
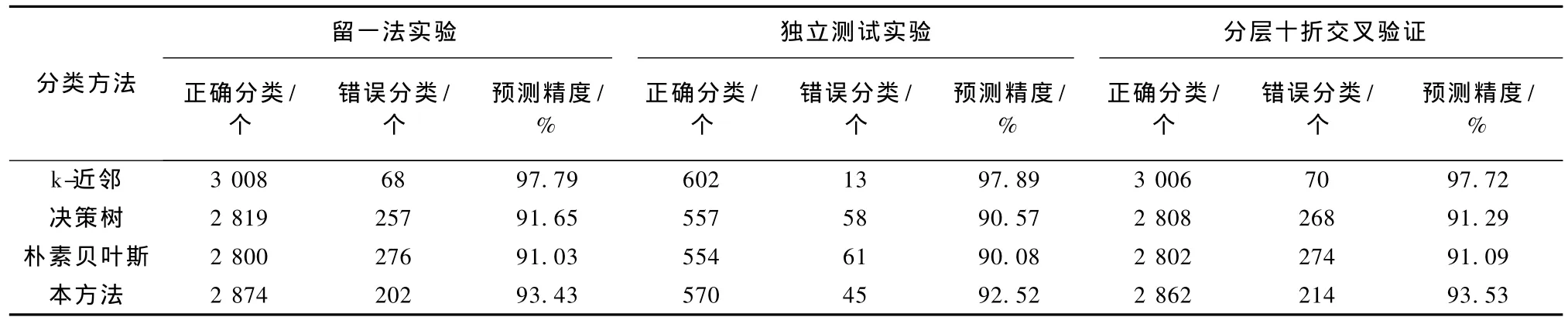
表3 本方法与k近邻,决策树和朴素贝叶斯方法的实验结果比较Tab.3 Comparision of the experiment results of the k-nearest neighour,decision treee,naive Bayes and the method of this paper
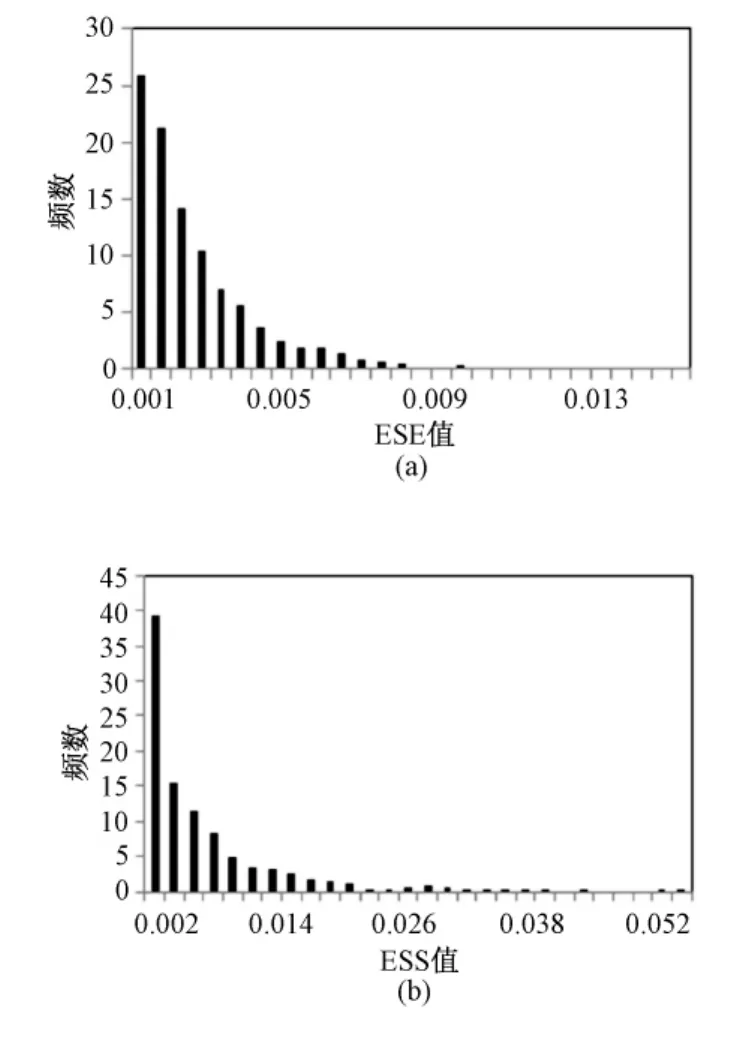
图3 PESE和PESS的剪接分值的统计直方图。(a)PESE的 ESEscore的统计直方图;(b)PESS的ESSscore的统计直方图Fig.3 Histogram for the splicing score of PESE and PESS.(a)Histogram for ESEscore of PESE;(b)Histogram for ESSscore of PESS
基于已知的ESE和ESS八联体,利用本方法计算所有未知八联体的倾向指数,从图2中,可以看到TR对ESE和ESS具有明确的标记功能,但在预测未知元件的剪接功能时,单独使用 TR也有不足。例如,一个未知元件具有高TR值,但是其ESEscore却较低,则其高 TR值的出现可能仅是由于其ESSscore更低造成的,因此,为了更准确地预测元件剪接功能,可以将TR值和剪接分值联合作为预测的依据。图3a和3b分别是 PESE的 ESEscore和PESS的ESSscore的统计直方图,从图中可以看出,超过 50%的 PESE的 ESEscore大于 0.002,超过50%的PESS的 ESSscore大于0.004。利用复合条件(TR>0.9 and ESEscore>0.002)和(TR<-0.9 and ESSscore>0.004)去预测高置信度的未知调控元件ESE和ESS,共获得555个新ESE和519个新ESS。表4和表5给出了部分预测结果。表4给出了部分具有高倾向指数和ESEscore的未知八联体,预测其具有ESE剪接功能,表5给出了部分具有低倾向指数和高ESSscore的未知八联体,预测其具有ESS剪接功能。本方法基于序列特征,计算未知元件的剪接分值,度量未知元件与已知元件的距离,从而预测未知元件的剪接功能,同时本方法具有透明的预测结构,还可以给出预测的依据,例如表4中,未知元件ccggagga的ESE剪接分值较高,而ESS剪接分值较低,则说明该元件更接近已知ESE,远离已知ESS,表现出较高的倾向指数,并且该元件包含多个ESE序列特征,与该未知元件具有相同序列特征的ESE八联体有ccggaggt和ccggacct等,这些都提供了预测未知元件剪接功能的生物依据。另外,比较表4和表5中所列出的部分ESE和ESS序列特征可以发现,ESE和ESS序列特征明显不同,从根本上来说,这种ESE和ESS序列特征的差异是由 ESE和ESS元件单核苷酸碱基成分构成的差异造成的。
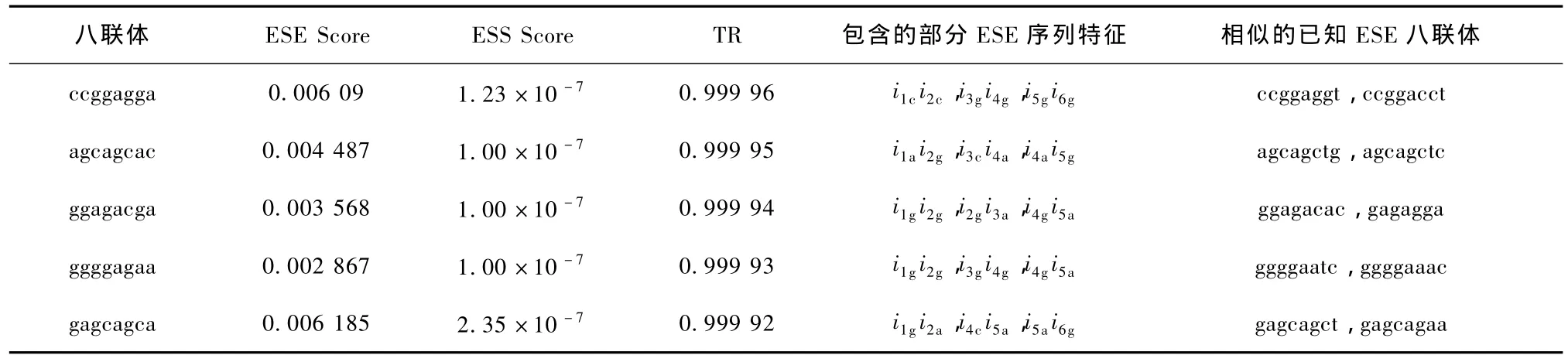
表4 预测的具有ESE功能的未知八联体Tab.4 Unkonwn octamers predicted with ESE function
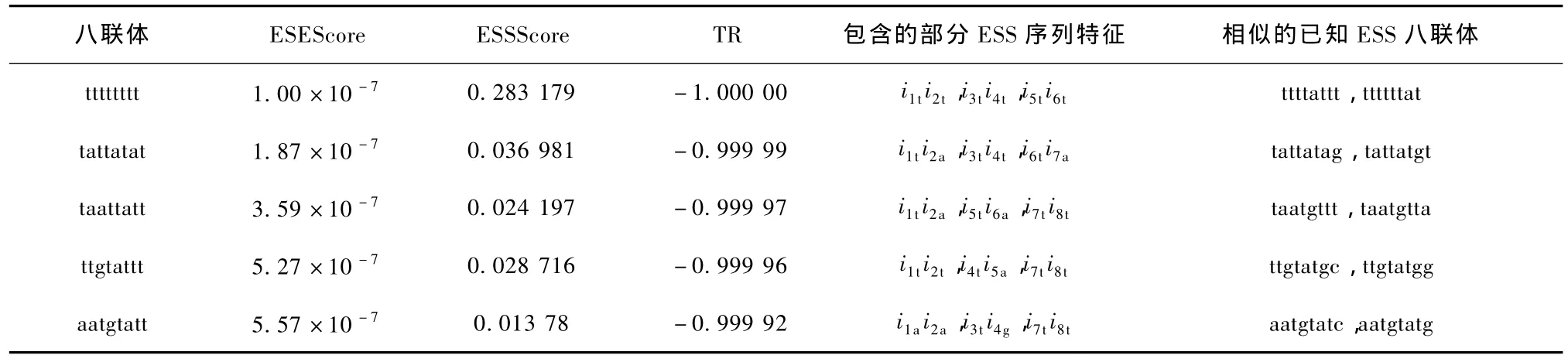
表5 预测的具有ESS功能的未知八联体Tab.5 Unkonwn octamers predicted with ESS function
上面实验说明了本方法分类识别剪接元件的有效性,但采用该方法进行分析时,其结果也会受其他因素的影响,这里考察不同的数据集规模以及采用不同的最小支持度时对数据分析结果的影响。
首先考察数据集规模对分析结果的影响。产生4对随机八联体数据集,仿照文中采用的ESS和ESE比例,每对数据集的规模比为1∶2,4对数据集规模分别为 (50∶100、150∶300、250∶500、750∶1500)。利用本方法求出这4对数据集的分值,分布结果如图4所示。由图4可以看出,随着数据集规模不断增大,其数据分类效果逐渐变差,究其原因,当两随机数据集规模较小时,其包含的序列特征差异较大,可以很容易将其分开,随着数据集规模增大,其共享的相似序列特征增多,两数据集的分值空间分布出现较大重叠,难以分开。从图4(d)可以看出,当随机数据集规模上千时,其分值空间分布几乎完全重叠,相较于图1,已知ESE和ESS清晰分开,这说明ESE和ESS包含着显著差异的序列特征。
采用不同的最小支持度,对同一序列数据计算所得的剪接分值不同,为了考察最小支持度对剪接分值计算的影响,这里本研究分别采用了四个不同的最小支持度:1%、4%、7%和10%,针对采用的ESE和ESS数据,计算剪接分值,空间分布结果如图5所示。由图5可以看出,当支持度取1%时,ESE和ESS清晰地聚为两簇,随着支持度增大,两簇逐渐发散,重叠区域增加,分类精度下降。当取较小支持度时,虽然数据分类效果较好,但易出现数据过拟合,为了获得较好的分类精度,同时避免数据过拟合问题,本研究采用了最小支持度5%。
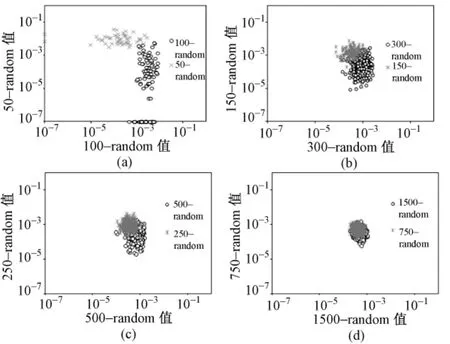
图4 不同数据规模的随机八联体数据集对剪接分值分布。(a)分别包含50和100个元件的两随机数据集;(b)分别包含150和300个元件的两随机数据集;(c)分别包含250和500个元件的两随机数据集;(d)分别包含750和1500个元件的两随机数据集Fig.4 Splicing score distribution for random octamers datasets with different scale.(a)Ttwo random datasets respectively including 50 and 100 elements;(b)Two random datasetsrespectively including 150 and 300 elements;(c)Two random datasetsrespectively including 250 and 500 elements;(d)Two random datasets respectively including 750 and 1500 elements
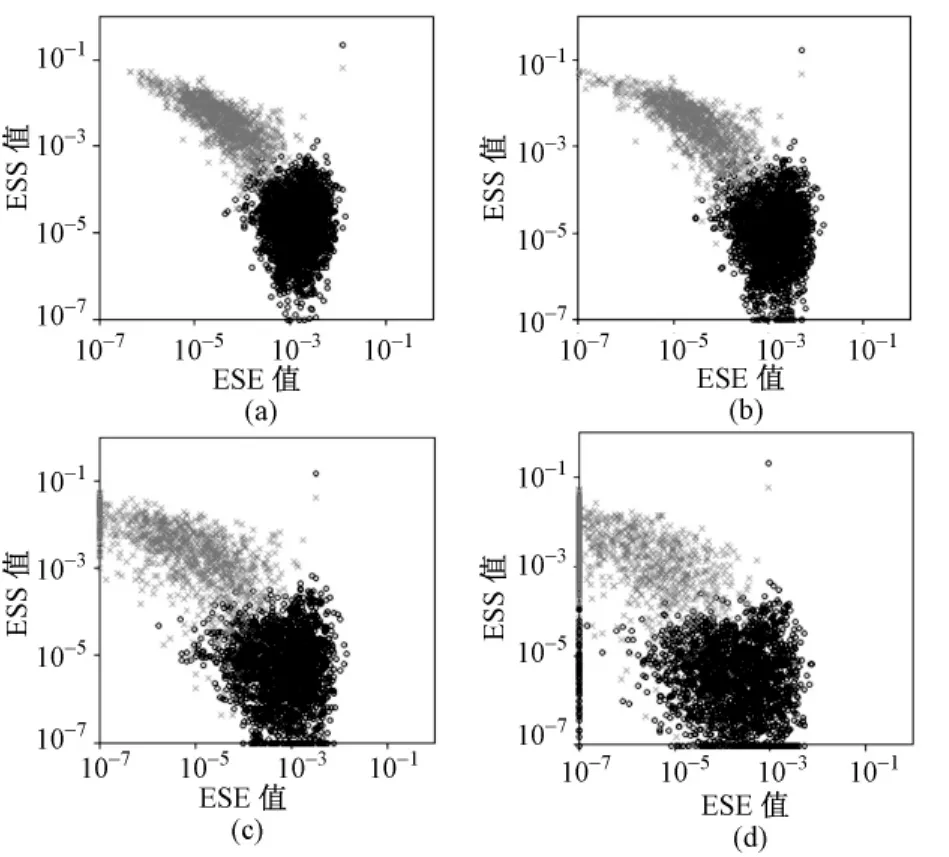
图5 基于不同的最小支持度时,已知ESE和ESS剪接分值分布。(a)基于最小支持度1%,已知ESE和 ESS剪接分值分布;(b)基于最小支持度4%,已知 ESE和ESS剪接分值分布;(c)基于最小支持度7%,已知 ESE和ESS剪接分值分布;(d)基于最小支持度10%,已知ESE和ESS剪接分值分布Fig.5 Splicing score distribution for the known ESE and ESS based on the different minsup.(a)Splicing score distribution for the known ESE and ESS based on minusp 1%;(b)Splicing score distribution for the known ESE and ESS based on minusp 4%;(c)Splicing score distribution for the known ESE and ESS based on minusp 7%;(d)Splicing score distribution for the known ESE and ESS based on minusp 10%
本方法也可被用于其他生物问题的研究。例如,SNP或突变对RNA剪接的影响。当某剪接元件内发生突变时,则该元件的剪接功能很可能会受到影响,或者增强,或者减弱,利用本方法,通过计算突变前后相应元件的剪接分值,可对突变的剪接影响进行定量分析。目前正在进行相关工作。
7 结论
RNA选择性剪接是导致人基因组生物复杂性的重要因素,与癌症、心血管疾病等发生密切相关。RNA剪接元件在RNA剪接过程中起着重要的调控作用,正确识别RNA剪接元件对深刻理解RNA剪接过程有着重要作用。本研究提供了一种基于已知剪接元件中的序列特征,计算未知元件的剪接分值,预测其剪接功能的方法,该方法简捷,具有良好的数理基础,计算验证实验结果表明,所提出的预测未知元件剪接功能的方法是可行的和有效的。
[1]Fairbrother WG,Yeh RF,Sharp PA,etal. Predictive identification of exonic splicing enhancers in human genes[J].Science,2002,297(5583):1007-1013.
[2]Zhang XH,Chasin LA.Computational definition of sequence motifs governing constitutive exon splicing[J]. Genes &Development,2004,18(11):1241 - 1250.
[3]Goren A.,Ram O,AmitM,etal. Comparativeanalysis identifies exonic splicing regulatory sequences--The complex definition of enhancers and silencers[J].Molecular Cell,2006,22(6):769-781.
[4]Wang Zefeng,Rolish ME,Yeo G,etal. Systematic identification and analysis of exonic splicing silencers[J].Cell,2004,119(6):831 -845.
[5]Arikawa S,Miyano S,Shinohara A,et al.A machine discovery from amino acid sequences by decision trees over regular patterns[J].New Generation Computing,1993,11(3):361 -375.
[6]Chuzhanova NA,Jones AJ,Margetts S.Feature selection for genetic sequence classification[J].Bioinformatics,1998,14(2):139-143.
[7]Blekas K,Fotiadis D,Likas A.Motif-based protein sequence classification using neural networks[J]. Journal of Computational Biology,2005,12(1):64 -82.
[8]Ratsch G,Sonnenburg S Schafer C.Learning Interpretable SVMs for BiologicalSequence Classification[J]. BMC Bioinformatics,2006,7(Suppl 1):S9.
[9]Leslie C,Kuang R.Fast string kernels using inexact matching for protein sequences[J].The Journal of Machine Learning Research,2004,5:1435 -1455.
[10]Sandberg R,Winberg G,Branden CI,et al.Capturing wholegenome characteristics in short sequences using a naive Bayesianclassifier[J].Genome Research,2001,11(8):1404 - 1409.
[11]Baldi P,Chauvin Y,Hunkapiller T,et al.Hidden Markov models of biological primary sequence information[J].Proceedings of the National Academy of Sciences of the United States of America,1994,91(3):1059 -1063.
[12]Li Haiquan,Dai Xinbin,Zhao Xuechun.A nearest neighbor approach for automated transporter prediction and categorization from protein sequences[J].Bioinformatics,2008,24(9):1129-1136.
[13]Wrzodek C,Schroder A,Drager A,et al.ModuleMaster:A new tool to decipher transcriptional regulatory networks.Biosystems,2010,99(1):79-81.
[14]Agrawal R,Srikant R.Fast algorithms for mining association rules[C] //Proceedings of 20thInternatinal Conference Cery Large Data Bases,VLDB.San Francisco:Morgan Kaufmann Publishers Inc,1994:487-499.
[15]Agrawal R,Imieli ski T,Swami A.Mining association rules between sets of items in large databases[C].Proceedings of the 1993 ACM Conference.Washington DC:ACM,1993:1-10.
[16]Aha DW,Kibler D,and Albert MK,Instance-based learning algorithms[J].Machine Learning,1991,6(1):37 - 66.
[17]Quinlan JR.C4.5:Programs for Machine Learning[M].SanMateo:Morgan Kaufmann Publishers,1993.
[18]John GH,Langley P.Estimating continuous distributions in Bayesian classifiers[C]// Proceedings of the Eleventh Conference on Uncertainty in Artificial Intelligence.San Mateo:Citesser,1995,1:338 -345.
