中国X连锁无丙种球蛋白血症40例基因型表型相关性分析
2012-01-05应文静孙金峤刘丹如俞晔珩王静漪王晓川
王 莹 应文静 孙金峤 刘丹如 俞晔珩 王静漪 王晓川
原发性低丙种球蛋白血症是原发性免疫缺陷病中最常见的一组疾病,其中X连锁无丙种球蛋白血症(XLA)是常见类型之一[1]。XLA是一种典型的B细胞缺陷病,绝大多数患者的临床表现为出生后不久即出现反复而严重的细菌感染、低丙种球蛋白血症及外周血B细胞的显著减少或缺乏[2]。然而多年的研究发现,XLA患者B细胞缺陷在分化发育中表现程度存在差异,因而反映在患者的临床表现也存在明显的差异。虽然研究表明XLA患者的平均确诊年龄是26个月,但仍有不少XLA患者直至成年后才得以诊断,且无明显的临床表现[3]。如何解释这种差异已成为近年来国际上关注的研究方向。
1993年,Vetrie等[4]通过定位克隆的方法找到了XLA的致病基因——Bruton′s酪氨酸激酶(BTK)基因 。截至2007年,根据国际BTK基因突变数据库的统计,全球已有620多种突变被报道,且没有一种突变的发生率超过3%[5]。目前,尚无研究证实XLA的疾病表型与基因型间存在明确的相关性,但XLA患者不同的BTK基因突变类型仍可能是影响疾病严重程度的因素之一[6,7]。2003至2011年,本研究通过对临床拟诊为原发性低丙种球蛋白血症患儿检测BTK蛋白表达和(或)BTK基因分析,确诊了40例XLA,并从确诊年龄、临床表现、免疫学功能、BTK蛋白表达及基因型等方面进行较为深入的分析,结果报道如下。
1 方法
1.1 病例来源 复旦大学附属儿科医院临床免疫科从2003年起建立了先天性免疫缺陷病的诊断流程,其中XLA的流程如下:对反复细菌感染、不明原因发热的临床怀疑XLA的患儿,行常规免疫功能评价(血清免疫球蛋白水平,T、B细胞数量),对临床拟诊XLA的患儿建立随访数据库(联系方式和临床资料的采集),进行至少包括患儿及其母亲的BTK基因分析,有条件者行流式细胞检测BTK蛋白表达[根据日本富山医科药科大学医学部小儿科 Miyawaki Toshio教授赠送抗BTK单克隆抗体(48-2H)的数量决定行这一检测的患儿数量]。
1.2 临床拟诊XLA标准 ①外周血CD19+的B细胞数量<2%;②血清免疫球蛋白水平<3 g·L-1;③反复较严重的细菌感染;④伴或不伴有XLA阳性家族史。
1.3 常规免疫功能评价 血清免疫球蛋白测定采用散射比浊法,使用免疫球蛋白TURBOX试剂盒(Orion Corpo-ration Orion Diagnostica, Espoo, Finland), T、B细胞分化簇(CD)测定使用荧光标记的抗CD19、抗CD3、抗CD4和抗CD8单克隆抗体 (BD Biosciences, San Jose, CA),结果采用Multiset软件分析。
1.4BTK基因突变检测BTK基因突变的检测方法根据Kanegane等[7]报道,简述如下:TRIzol (Invitrogen Corp, Carlsbad, CA)抽提外周血单个核细胞(PBMC)总RNA,5 μg总RNA Invitrogen ThermoSciptTM使用 RT-PCR System (Invitrogen Life Tech-nologies, NY,USA)作用下逆转录获得cDNA。使用8对重叠的引物分2步扩增BTKcDNA。使用QIAGEN血液DNA抽提试剂盒(QIAGEN,USA)基因组DNA,一系列根据BTK基因外显子序列设计的引物用于DNA PCR。PCR产物经碱性磷酸酶(SAP)(Fermentas, USA)纯化后直接测序。测序反应使用BigDye Terminator Cycle Sequencing 试剂盒(Applied Biosystems, Foster City, CA),产物经纯化后使用ABI PRISM 3130 DNA测序仪(Applied Biosystems)进行测序。由cDNA PCR产物测序发现的BTK突变,经相对应部位的DNA 序列PCR产物进行测序进一步证实。患儿母亲及家族中部分亲属DNA也在相应部位扩增并测序。
1.5 流式细胞仪检测单核细胞BTK蛋白表达 采集患儿及其家庭成员肝素抗凝的外周血,Ficoll-Hypaque (Sigma, USA) 密度梯度离心分离外周血单个核细胞。使用48-2H标记细胞内BTK[8]。具体步骤:使用PE标记的抗CD14 (IgG2a; DAKO, Kyoto, Japan) 单克隆抗体加入PBMC,冰浴20 min。4%多聚甲醛室温固定细胞15 min。0.1%TBS Triton X-100 (pH 7.4)含0.1%BSA破膜5 min。加入2 μg·mL-148-2H或IgG1 (DAKO)单克隆抗体,冰浴20 min;洗涤后加1∶2 000稀释的FITC标记的羊抗小鼠IgG1 (Southern Biotechnology Associates, Birmingham, AL),冰浴20 min。流式细胞仪采用CellQuest软件采集并分析结果。
2 结果
2.1 一般情况 2003年1月至2011年5月复旦大学附属儿科医院临床免疫科共连续收集来自47个家族的50例男性原发性低丙种球蛋白血症患儿。40例确诊XLA患儿起病年龄、确诊年龄和免疫表型见表1。
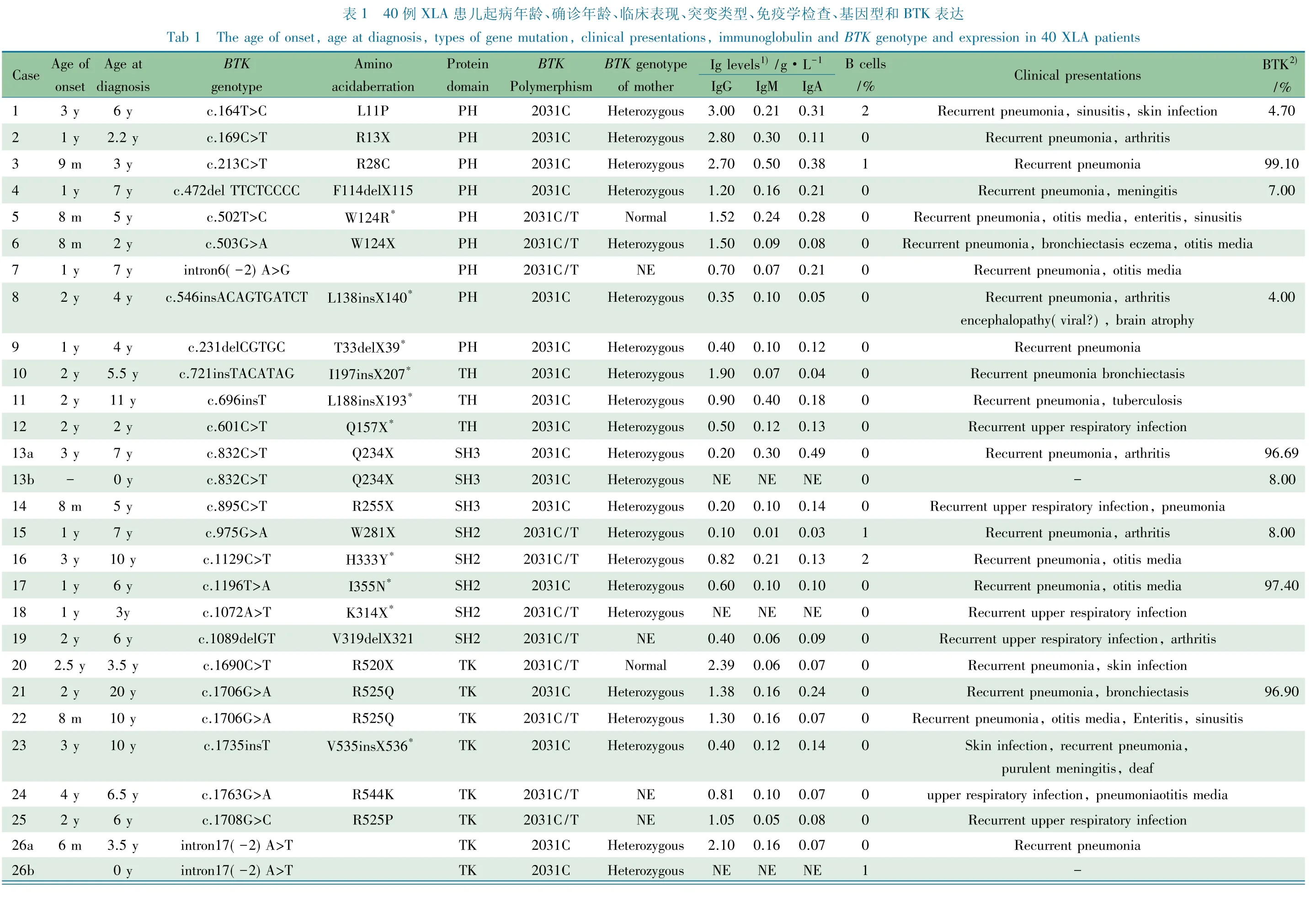
2.2 XLA患儿BTK基因突变类型及特点 50例临床拟诊为原发性低丙种球蛋白血症患儿行基因分析,其中40例(80%)BTK基因存在突变,确诊为XLA,14/40例为未见报道(http://www.uta.fi/imt/bioinfo/BTKbase/)的新突变(表1)。40例基因突变包括:16例(40.0%)错义突变,13例(32.5%)无义突变,4例(10.0%)插入突变,4例(10.0%)缺失突变和3例(7.5%)剪接突变。8例位于CpG位点(R13X、R28C、R255X、R520X、R525P、R562L各1例,R525Q 2例)。40例基因突变分布于整个BTK基因编码区域,其中20例(50.0%)位于激酶(TK)区,9例(22.5%)位于PH区,3例(7.5%)位于TH区,3例(7.5%)位于SH3区,5例(12.5%)位于SH2区(表2,图1)。21/40例(52.5%)被证实第18号外显子2031位碱基存在C/T多态性,其他BTK基因数据库中报道的多态性未在本研究对象中检测到(表1)。
表2 40例XLA患儿BTK基因突变类型及结构域分布[n(%)]
Tab 2BTKmutation types and distribution in BTK protein domain[n(%)]
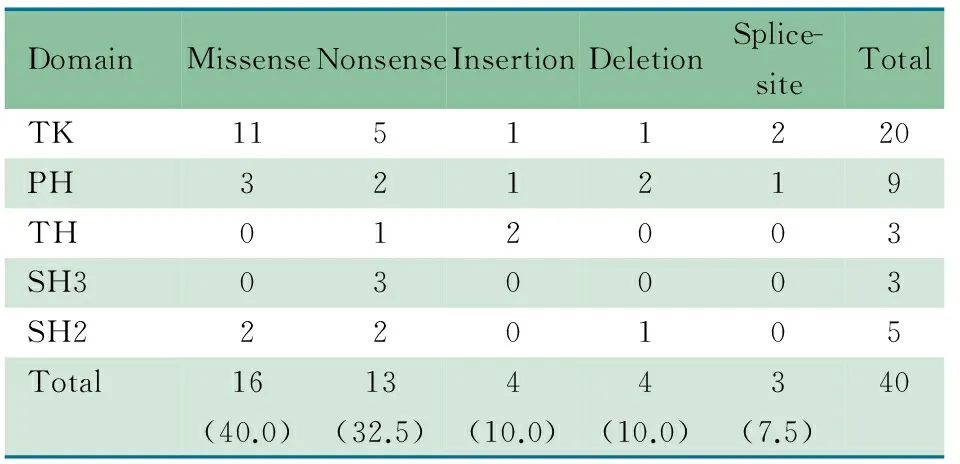
DomainMissenseNonsenseInsertionDeletionSplice-siteTotalTK11511220PH321219TH012003SH3030003SH2220105Total16(40.0)13(32.5)4(10.0)4(10.0)3(7.5)40
Notes PH: pleckstrin homology;TH :Tec homology;SH:Src homology; TK:Tyrosine Kinase
2.3 临床表现、确诊年龄与基因型关系 表1显示,XLA患儿均有典型的原发性低丙种球蛋白血症的临床表现,即反复发生的细菌性感染,包括肺炎、中耳炎、腹泻、骨髓炎、脑膜炎、脊髓灰质炎及皮肤感染。9例(例2,8,13,15,19,31,33,34a和34b)伴有关节炎。例22还伴发颜面及球结膜毛细血管扩张。
40例确诊XLA患儿起病年龄均≤4岁,24例起病≤1岁。突变类型为错义突变的患儿平均起病年龄为(1.4±1.1)岁,其他突变类型患儿为(1.4±0.7)岁,差异无统计学意义(P=0.45)。
40例XLA患儿确诊年龄与BTK基因突变类型相关性如表3所示,确诊年龄≤3岁9例,~6岁15例,~10岁13例,>10岁3例。结果显示,错义突变的发生率随年龄的增长呈上升趋势,无义突变的发生率呈下降趋势。≤3岁患儿中错义突变2例(1例位为编码PH区第28位氨基酸的CpG位点,1例位于TK区),1例为位于TK区的剪接突变。例21和27起病年龄均为2岁,诊断年龄例21为20岁,例27为13岁,均为位于TK区CpG位点的错义突变。
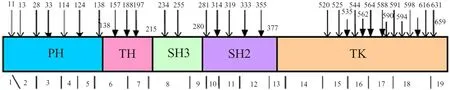
图1 XLA患儿BTK基因突变类型及分布
Fig 1BTKgene mutation types and distribution in protein domain
NotesBTKgene contains 19 exons. BTK protein contains five function domains. All forty cases ofBTKmutation were marked in the figure. Mutation marked with solid black arrows were new types that had not been reported in the BTKbase.PH: pleckstrin homology;TH :Tec homology;SH:Src homology; TK:Tyrosine Kinase. There were 9, 3, 3,5 ,20 cases in PH, TH SH3 SH2 and TK domains
表3 XLA患儿确诊年龄与BTK基因突变类型[n(%)]
Tab 3 Age at diagnosis and type ofBTKgene mutation in XLA patients[n(%)]
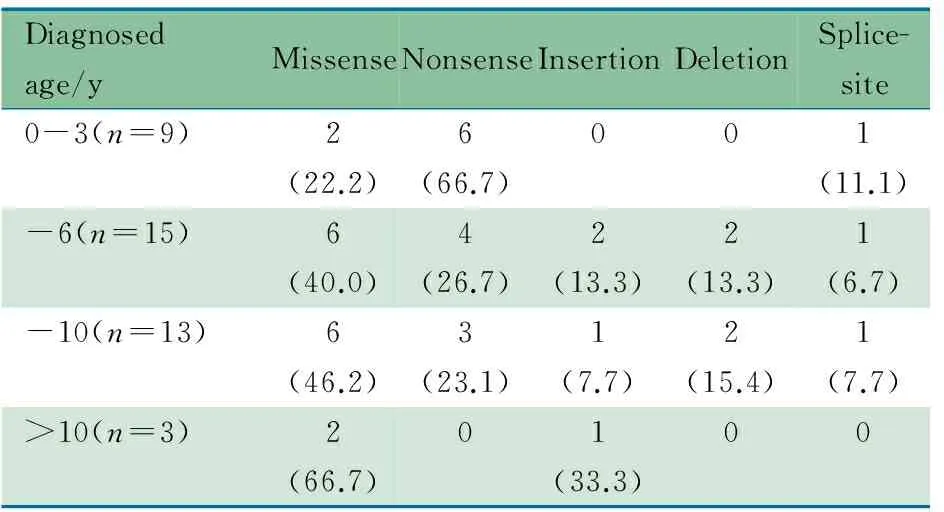
Diagnosedage/yMissenseNonsenseInsertionDeletionSplice-site0-3(n=9)2(22.2)6(66.7)001(11.1)-6(n=15)6(40.0)4(26.7)2(13.3)2(13.3)1(6.7)-10(n=13)6(46.2)3(23.1)1(7.7)2(15.4)1(7.7)>10(n=3)2(66.7)01(33.3)00
2.4 常规免疫功能水平与基因型关系 40例确诊XLA患儿常规免疫功能评价均有外周血B细胞显著减少或缺乏,其中错义突变13例(32.5%),无义突变12例(30.0%)。34/40例(85.0%)B细胞<0.1%;4例(10.0%)B细胞在1%~2%,其中错义突变2例,无义突变1例,剪接突变1例;2例(5.0%)B细胞为2%,均为错义突变(表1)。
37/40例XLA患儿检测了血清IgG水平,其中<1 g·L-120例,~3 g·L-116例, >3 g·L-11例。表4显示,血清IgG<3 g·L-1患儿BTK基因突变类型以错义突变和无义突变为主,且突变大多位于TK区(表1)。
例33血清IgG<0.1 g·L-1,其基因突变类型为位于TK区的错义突变(L616F);例27血清IgG>4 g·L-1,其基因突变类型为位于TK区CpG位点的错义突变(R562L)(表1)。基因突变类型为错义突变和其他突变类型患儿血清IgG水平分别为(1.43±1.15)和(1.05±0.81)g·L-1,差异无统计学意义(P=0.25)。
表4 XLA患儿血清IgG水平与BTK基因突变类型[n(%)]
Tab 4 Serum IgG level and type ofBTKgene mutation in XLA patients[n(%)]
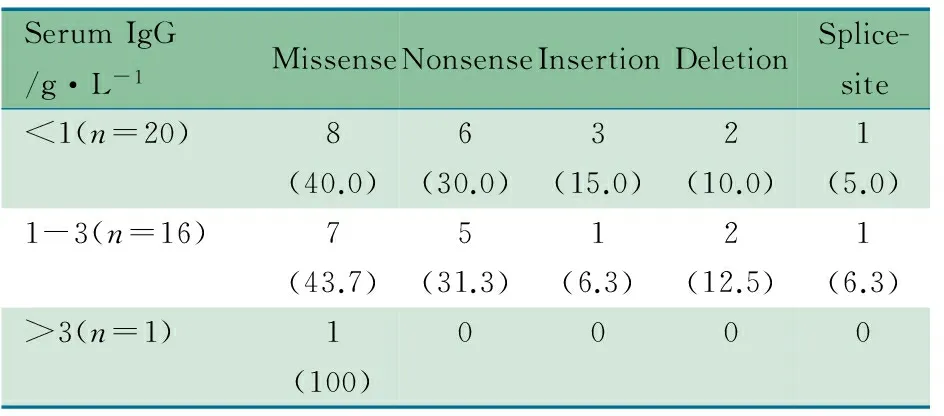
SerumIgG/g·L-1MissenseNonsenseInsertionDeletionSplice-site<1(n=20)8(40.0)6(30.0)3(15.0)2(10.0)1(5.0)1-3(n=16)7(43.7)5(31.3)1(6.3)2(12.5)1(6.3)>3(n=1)1(100)0000
2.5 单核细胞BTK蛋白表达水平与基因型关系 13例XLA患儿检测了单核细胞BTK蛋白表达(表1),7/13例(53.8%)BTK蛋白表达明显降低,其中错义突变3例(42.8%),无义突变2例(28.6%),插入突变和缺失突变各1例(14.3%)。余6例单核细胞BTK蛋白表达正常,其中4例(66.7%)突变类型为错义突变。错义突变患儿BTK蛋白表达水平为(49.9±45.1)%,其他突变类型为(52.4±50.1)%,差异无统计学意义(P=0.16)。例3的BTK蛋白表达水平正常,为位于PH区的CpG位点的错义突变,血清IgG水平为2.9 g·L-1,较其他患儿高(表1)。采用Spearman等级相关分析,XLA患儿单核细胞BTK蛋白表达水平与外周血B细胞数量(r=0.287)和血清IgG水平(r=0.35)无显著相关性。
2.6BTK基因多态性与疾病表型 6/21例(28.6%)2031位碱基存在C/T多态性患儿(例2、8、31、33、34a和34b)临床表现除了上呼吸道反复感染外还伴有严重的关节炎,主要表现为关节损伤、变形;19例无多态性患儿中3例(15.8%)有关节炎表现。
2.7 家族遗传特点分析 32例XLA患儿母亲检测了BTK基因突变,其中28例(87.5%)患儿母亲BTK基因为杂合型,证实为携带者(表1)。余4例患儿母亲基因分析正常,其基因突变可能为新发突变(denovomutation)。
3 讨论
中国从1998年开始原发性免疫缺陷病登记工作[9],国内有关XLA的报道多数为临床诊断病例。Pamela等[10]报道了包括中国(大陆和香港)和新加坡在内的62例基因确诊病例,本课题组前期报道了30例XLA病例[11]。本研究采用BTK基因分析结合流式细胞法检测单核细胞内BTK蛋白表达,确诊了40例XLA[12~14]。本研究结果显示,中国XLA患儿主要的突变类型为错义突变,突变主要分布于TK区和PH区,而这两个结构域也是BTK蛋白中最主要的功能区。根据国际BTK基因突变数据库的统计信息,本研究结果与全球BTK基因突变类型及在BTK基因中的分布相近[15]。
Pamela等[10]对62例XLA患者的起病年龄进行研究后发现,突变类型为错义突变的患者起病年龄明显高于其他突变类型的患者,而本研究错义突变患儿的起病年龄与其他突变类型无显著差异。由于不同地区的环境及医疗条件的差异,部分XLA患儿在初次发病后未能得到及时的诊断,因而造成了本研究中XLA患儿起病年龄和确诊年龄的差异。有研究表明XLA患者的平均确诊年龄为26个月[3],本研究40例XLA患儿平均确诊年龄为5.5岁(66月龄),确诊年龄在3岁以下患儿以无义突变为主,确诊年龄在6岁以上患儿以错义突变为主。这与国际上一些大样本的研究结果一致[10,16]。Broides等[17]诊断110例XLA患儿中有56%诊断年龄在6岁以上者的基因突变类型为错义突变,且随着诊断年龄的延迟,错义突变在患儿中所占的比例也逐渐升高。因此,结合国际上的大样本研究可见XLA患儿的突变类型可能与疾病的确诊年龄有关,而引起单个氨基酸替代改变的错义突变可能是引起患儿确诊年龄延迟的因素之一。起病年龄一般认为可以反映疾病的严重程度,但由于是回顾性病史询问,较难客观。而确诊年龄受到包括医疗条件、经济条件、认知度以及疾病严重程度等多方面的影响,也可以一定程度反映疾病的严重程度。
XLA患儿外周血成熟B细胞水平与血清免疫球蛋白水平是评价疾病严重程度的重要指标之一。有研究发现,能引起蛋白结构严重改变的突变类型如无义突变、插入突变或缺失突变的XLA患儿常规免疫功能水平并无明显低下[18]。本研究错义突变XLA患儿血清IgG水平与其他突变类型无显著差异。但结合国内外其他大样本及本研究结果提示,错义突变者的临床改变较轻,血清IgG水平相对较高。
近年来的研究发现,真核生物甲基化位点,CpG位点是BTK基因序列中较易发生突变的位点。在BTK整个编码序列中共有18个CpG位点,而其中仅有8个位点(13,28,255,288,520,525,562和641)有突变报道[12]。本研究中有8例XLA患儿突变位于CpG位点,1例突变位于PH区的编码第28位氨基酸的CpG位点。从BTK蛋白结构来看,位于PH区的第28位精氨酸是重要的磷脂酰肌醇结合位点,也是BTK蛋白介导信号转导途径中重要的结构位点,因此该位点突变无论何种突变类型均会造成蛋白结构改变,导致蛋白功能障碍[19]。但也有研究指出,位于该位点的错义突变(R28C)并不会造成患者出现严重的临床表现,且仍保留部分正常的BTK蛋白表达[20]。本研究例3的血清IgG水平和外周血B细胞数量均高于其他患儿,且临床表现相对较轻。另2例错义突变位于TK区的第525位精氨酸(CpG)位点,该位点无论是错义突变或无义突变均可能导致明显的疾病临床表现[21],其中例22临床表现较特殊,除反复呼吸道感染外还伴有颜面部的毛细血管扩张,是否与CpG位点突变类型有关尚不清楚;例2突变位于PH区的编码第13位氨基酸的CpG位点,这一位点的无义突变亦可造成蛋白结构的改变导致蛋白的功能障碍。
位于PH结构域上的L11至T33包含众多的保守序列,氨基酸F10-X-K12-S14-X10-F25-K26-X-R28-X-F30-X-L32-X4-L37-X-Y39构成PH结构域中重要的3-磷酸肌醇结合位点,其中F10、L31、Y39、F102、V113 和I125是高度保守的氨基酸残基,这些位点突变直接影响BTK蛋白的功能,可出现较为典型的XLA临床表现[13]。本研究中3例基因突变位于该区域,其中例1和3为错义突变(L11P和R28C),例2为无义突变(R13X),且血清IgG水平均高于2 g·L-1,例3 BTK蛋白表达水平明显高于例1。研究表明,位于PH结构域的突变均能造成该结构与磷酸肌醇结合的亲和力下降,而某些位点如R28的突变则仍保留较高的与磷酸肌醇结合的亲和力,因而所造成的临床表现也较轻[13,20]。本研究中有8例基因突变位于SH3和SH2结构域,其中有2例为错义突变(H333Y和I355N),为BTK基因突变数据库中尚未报道的突变类型。值得注意的是,错义突变可以发生于BTK基因除了TH结构域中的脯氨酸富集区及整个SH3区的各个结构域[18,22]。这可能与SH3结构域在BTK蛋白功能中的负调控功能有关。本研究3例位于SH3结构域的突变均为无义突变。
研究表明,SH3和SH2结构域与TK区和PH区相互作用,某些错义突变可能只改变蛋白的功能而不影响蛋白的空间结构[18]。本研究例17位于SH2结构域的错义突变(I355N),可检测到正常的BTK蛋白表达。研究表明,SH2结构域中的R288和V319是BTK蛋白中重要的保守位点,这些位点发生的突变可能干扰BTK蛋白与配体的结合[18]。本研究例19位于V319的插入突变,其外周血B细胞缺如,血清IgG<1 g·L-1,有较为典型的临床表现,且伴有严重的关节炎。
XLA患儿单核细胞BTK蛋白的表达水平也是反映疾病严重程度的指标之一。本研究中所使用的48-2H是针对BTK基因的SH3功能区,对其他功能区发生的突变也可检测到BTK的表达异常,但可能存在BTK蛋白表达程度差异[23]。本研究7/13例(53.8%)BTK蛋白表达明显降低,以引起BTK蛋白结构明显改变的无义突变、插入突变及缺失突变为主,而BTK蛋白表达正常患儿以错义突变为主。提示,不同的基因突变类型与BTK蛋白表达水平密切相关故而影响疾病的表型,但未检测到BTK蛋白表达水平降低并不能排除BTK基因突变的存在。本研究结果显示,单核细胞BTK蛋白表达的多少与外周血B细胞的数量以及血清IgG水平无显著相关,这与文献报道较为一致,具体原因还不清楚,可能是基因型与表型间还存在多种其他因素的影响[6,7,10,22]。
XLA患者家系中女性携带者的判定对于优生有重要的意义。本研究对32例患儿母亲及部分家族亲属进行了基因分析,其中28例母亲携带有突变的BTK基因,4例患儿母亲基因分析正常,其基因突变可能是卵母细胞新发生的突变[8]或生殖腺嵌合型所致。Parolini等[24]报道首例XLA患儿母亲为生殖腺嵌合型。本研究提示携带BTK基因突变女性亲属可能将所携带的突变基因遗传给下一代,且XLA患儿母亲再次分娩带有突变基因XLA患儿的风险较高。
XLA的合并症与BTK基因间的关系尚不清楚。XLA中最重要的一类合并症是关节炎并可致残。本组患儿关节炎的发生率为22.5%(9/40例)。进一步分析显示,21例第18号外显子2031位碱基存在C/T多态性XLA患儿关节炎的发生率为28.6%(6/21例),高于无多态性患儿(15.8%,3/19例),这是否提示多态性的存在可能增加发生关节炎风险还有待进一步研究。
目前,虽然尚无有力的证据证实XLA患儿基因型与表型之间存在明确关系,但通过本研究并结合国外较大样本的分析可见,仅引起单个氨基酸替代改变的错义突变可能与较大的诊断年龄、较高的外周血B细胞数量和血清IgG水平以及与正常的BTK蛋白表达水平有关。本研究中21例XLA患儿存在BTK基因多态性(2031C/T),这种多态性可能增加合并关节炎的风险。
注:本文部分基因突变数据发表于J Clin Immunol,2009,29(3):352-356
[1]Bruton OC. Agammaglobulinemia. Pediatrics, 1952, 9(6): 722-728
[2]Wang XC(王晓川). Clinical features of X-linked agammaglobulinemia:analysis of 8 cases. Chin J Pediatr(中华儿科杂志), 2004, 42(8): 564-567
[3]Conley ME, Howard V. Clinical findings leading to the diagnosis of X-linked agammaglobulinemia. J Pediatr, 2002, 141(4):566-571
[4]Vihinen M, Kwan SP, Lester T, et al. Mutations of the human BTK gene coding for Bruton Tyrosine kinase in X-linked agammaglobinemia. Human mutation, 1999, 13(4):280-285
[5]Conley ME, Broides A, et al. Genetic analysis of patients with defects in early B cell development. Immunol Rev, 2005, 203(1): 216-234
[6]Teimourian S, Nasseri S, Pouladi N, et al. Genotype-phenotype correlation in Bruton′s tyrosine kinase deficiency. J Pediatr Hematol Oncol, 2008, 30(9): 679-683
[7]Kanegane H, Futatani T, Wang Y, et al. Clinical and mutational characteristics of X-linked agammaglobulinemia and its carrier identified by flow cytometric assessment combined with genetic analysis. J Allergy Clin Immunol, 2001,108(6): 1012-1020
[8]Plebani A, Soresina A, Rondelli R, et al. Clinical, immunological and molecular analysis in a large cohort of patients with X-linked agammaglobulinemia: An Italian multicenter study. Clin Immunol, 2002, 104(3):221-230
[9]中华医学会儿科学分会, 中华儿科杂志编辑委员会. 原发性免疫缺陷病的协作网和登记工作. Chin J Pediatr(中华儿科杂志), 1999, 37(6):328-329
[10]Lee PP, Chen TX, Jiang LP, et al. Clinical characteristic and genotype-phenotype correlation in 62 patients with X-linked agammaglobulinemia. J Clin Immunol, 2010, 30:121-131
[11]Wang Y, Kanegane H, Wang X, et al. Mutation of the BTK gene and clinical features of X-linked agammaglobulinemia in mainland China. J Clin Immunol, 2009, 29(3):352-356
[12]Lindvall JM, Blomberg KE, Voliaho J, et al. Bruton′s tyrosine kinase: cell biology, sequence conservation, mutation spectrum, siRNA modifications, and expression profiling. Immunol Rev, 2005, 203: 200-215
[13]Shen B, Vihinen M. Conservation and covariance in PH domain sequences: physicochemical profile and information theoretical analysis of XLA causing mutations in the BTK PH domain. Protein Eng Des Sel, 2004,17(3):267-276
[14]The Protein Kinase Resource: the enzymology, genetics, molecmolecular and structural properties of protein kinases. Available from http://www.nih.go.jp/mirror/Kinases/pk_home.html
[15]Vihinen M, Belohradsky BH, Haire RN, et al. BTKbase, mutation data base for X-linked agammaglobulinemia. Nucleic Acids Res,1997, 25(1): 166-171
[16]Holinski-Feder E, Weiss M, Brandau O, et al. Mutation screening of the BTK gene in 56 families with X-linked agammaglobulinemia (XLA): 47 unique mutations without correlation to clinical course. Pediatrics, 1998, 101(2): 276-284
[17]Broides A, Yang W, Conley ME. Genotype/phenotype correlations in X-linked agammaglobulinemia. Clin Immun, 2006, 118(2-3): 195-200
[18]Lappalainen I, Thusberg J, Shen B, et al. Genome wide analysis of pathogenic SH2 domain mutations. Proteins, 2008, 72(2):779-792
[19]Wang XC(王晓川). Juvenile idiopathic arthritis in patients with X-linked agammaglobulinemia. Shanghai Med J(上海医学), 2003, 26(7): 470-472
[20]Kojima T, Fukuda M, Watanabe Y, et al. Characterization of the pleckstrin homology domainof Btk as an inositol polyphosphate and phosphoinositide binding domain. Biochem Biophys Res Commun, 1997, 236(2): 333-339
[21]Mao C,Zhou M,Uckun FM. Crystal Structure of Bruton′s tyrosine kinase domain suggests a novel pathway fox activation and provides insights into the molecular basis of X -linked agammaglobulinemia. J Biol Chem,2001,276(44):41435-41443
[22]López-Granados E, Pérez de Diego R, Ferreira Cerdán A, et al. A genotype-phenotype correlation study in a group of 54 patients with X-linked agammaglobulinemia. J Allergy Clin Immunol, 2005, 116(3):690-697
[23]Wang XC(王晓川), Yu YH. Expression of Bruton′s tyrosine kinase in primary hypogammaglobulinemia. Chin J Microbiol Immunol(中华微生物学和免疫学杂志), 2004, 24(2):121-124
[24]Parolini O, Hejtmancik JF, Allen RC, et al. Linkage analysis and physical mapping near the gene for X-linked agammaglobulinemia at Xq22. Genomics , 1993, 15(2):342-349
