郑氏比蜢线粒体基因组全序列的测定与分析
2011-12-25杨辉,黄原
杨 辉, 黄 原
(陕西师范大学 生命科学学院, 陕西 西安 710062)
郑氏比蜢线粒体基因组全序列的测定与分析
杨 辉, 黄 原∗
(陕西师范大学 生命科学学院, 陕西 西安 710062)
采用长距PCR 扩增及保守引物步移法测定并注释了郑氏比蜢(Pielomastax zhengi)的线粒体基因组全序列。郑氏比蜢的线粒体基因组全长15 602 bp, A+T 含量为71.8%, 37个基因位置与飞蝗的一致, 基因间隔序列共计10处47 bp, 间隔长度从1~20 bp不等; 有14对基因间存在52 bp重叠, 重叠碱基数在1~8 bp之间。蛋白质基因的起始密码子均为昆虫典型的起始密码子ATN。ND5 基因使用了不完全终止密码子T, 其余基因均为典型的TAA或TAG。除 tRNASer(AGN)的 DHU 臂缺失外, 其余21个tRNA 基因的二级结构均属典型的三叶草结构, 但在郑氏比蜢中有5个tRNA(tRNACys、tRNALys、 tRNAPhe、 tRNAProtRNAArg)基因变异较大, 无法采用常规算法预测出来, 表现在这5个tRNA 二级结构的TψC臂仅有3~4对配对碱基, tRNALys和tRNAArg的反密码臂仅有4对配对碱基。预测的 lrRNA 二级结构总共有 6 个结构域(结构域Ⅲ缺失), 44 个茎环结构。预测的 srRNA 的二级结构包含3 个结构域, 30 个茎环结构。比较郑氏比蜢、西藏飞蝗(Locusta migratoria tibetensis)和疑钩额螽(Ruspolia dubia)rRNA二级结构后,发现郑氏比蜢与西藏飞蝗的更相似。A+T 丰富区中存在一个被认为与复制及转录起始有关的 Ploy(T)结构。
蜢总科; 郑氏比蜢; 线粒体基因组; RNA二级结构
后生动物线粒体基因组通常是编码 37个基因(13个蛋白质基因、22个tRNA基因及2个rRNA基因)和1个控制区(也称A+T丰富区)的细胞器基因组(Wolstenholme, 1992; Ding et al, 2007)。线粒体DNA具有多拷贝数、母系遗传、突变率高、极少发生重组等特性, 已被广泛地运用于分子进化、系统地理学和系统发育关系等方面的研究(William et al,2001)。根据 NCBI(www.ncbi.nlm.nih.gov/genomes/ORGANELLES/mztax_short.html)的统计, 截至2010年 11月, 已测出全线粒体基因组的昆虫纲种类有223种, 其中直翅目昆虫34种。由于测序的类群相对较少, 所选物种的特殊进化模式及系统树中可能的长枝吸引等问题, 利用线粒体基因组数据推断直翅目系统发育和进化地位还存在一定的困难(Gao et al, 2009)。另外, 已测的34种直翅目昆虫大部分是螽斯总科和蝗总科的, 还缺乏蜢总科的代表种类, 因而增加这些类群中有关线粒体基因组全序列数据将有利于建立比较稳健的系统发育关系。
郑氏比蜢(Pielomastax zhengi)属于直翅目蜢总科(Eumastacoidea)枕蜢科(Episactidae)。蜢总科是一类较罕见而又原始的小型昆虫, 其中一些种类还被认为是第三纪的孑遗种类。它们的大多数种类生活在热带和亚热带的灌木丛或林区内(Huang et al,2009)。本文测定并注释了郑氏比蜢线粒体基因组全序列, 并对其编码的22个tRNA和大小亚基rRNA的二级结构做了预测和分析, 以期为直翅目昆虫线粒体谱系基因组学提供新的数据资料。
1 材料和方法
1.1 标本采集及总DNA的提取
郑氏比蜢标本于2009年8月采集于河南内乡县宝天曼自然保护区, 保存于无水乙醇。总DNA采用酚-氯仿抽提法:将后足股节肌肉置于 600 µL 匀浆缓冲液(0.01 mol/L Tris, pH 7.8; 5 mmol/L EDTA,5% SDS, 50 ng proteinase K/µL)中研磨, 65 ℃水浴消化 3~5 h。用平衡酚(pH 7.6~7.8)提纯2次, 再用 CI(氯仿∶异戊醇=24∶1)提纯一次。最后用−20 ℃预冷的 100%乙醇沉淀和 70%乙醇洗涤总DNA, 保存于−20 ℃。
1.2 PCR扩增及测序
本研究所采取的测序策略是长 PCR产物步移法测序, 如有测序效果不佳或不能够测通的区域,再做 sub-PCR来补齐缺口。首先, 通过四对长距PCR(L-PCR)引物将线粒体基因组扩增为 4条相互重叠的片段(片段A、A2、B1和B2), 4个片段的上下游引物序列见表1(Liu et al, 2006), 长距PCR反应体系的总体积是 15 μL, 包括 10×反应缓冲液 1.5 μL、25 mmol/L MgCl22 μL、2.5mmol/L dNTPs 3 μL,10 μmol/L 上、下游引物各 2μL、5 U/μL LA-TaqDNA聚合酶0.15~0.35 μL、总DNA为模板1 μL。然后,加ddH2O至终体积15 μL。PCR反应循环条件如下:93 ℃预变性 2 min→(92 ℃10 s, 48 ℃ 30 s, 68 ℃8 min)共 15 个循环→(92 ℃ 10 s, 48 ℃30 s, 68 ℃8 min +20 s)共 15个循环→68 ℃ 7 min→4 ℃保存。其次, 用 12对保守引物以片段 A、A1、B1和 B2为模板再扩增320~3 175 bp的短片段, 具体信息见表2 (Simon et al, 2006)。短片段扩增体系总体积为25 μL, 包括 10×反应缓冲液 2.5 μL、25 mmol/L MgCl23 μL、2.5 mmol/L dNTPs 2 μL, 10 μmol/L 上、下游引物各4 μL、5 U/μL TaqDNA聚合酶0.15~0.35 μL和长片段产物(作为DNA模板)1 μL。然后,加ddH2O至终体积25 μL。PCR 反应程序为: 94 ℃2 min→(94℃×30 s+38~55 ℃×30 s+72 ℃×1 min)共 30 个循环→72 ℃×7 min→4 ℃ forever。最后, 通过12条sub-PCR产物测序将缺口补齐, 使基因组连接成环。

表1 长PCR扩增引物序列Tab. 1 Primer sequences for long PCR amplication
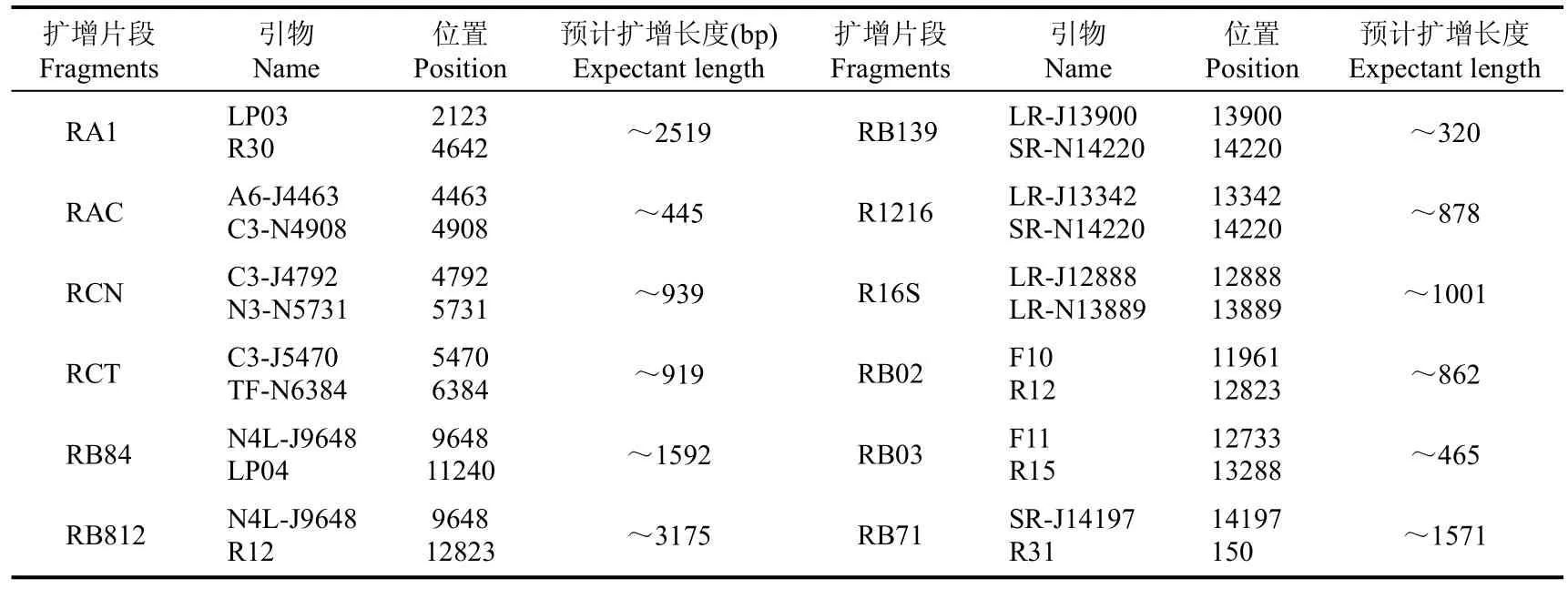
表2 补充扩增片段的引物对Tab. 2 The partial pair primers for the mitogenome complement fragment amplification
1.3 序列拼接、注释和分析
使用Staden Package 1.7 (Bonfield et al, 1995)进行序列拼接和注释, 通过与威廉剑角蝗Acrida_willemsei(NC_011303)线粒体 DNA序列进行比对确定蛋白质基因、tRNA基因和rRNA基因的位置,使用 MEGA 4.0 统计线粒体基因组碱基组成、蛋白质基因密码子使用频率等。使用 tRNAScan-SE ver.1.21进行 tRNA 二级结构预测。参考已发表的李氏大足蝗 srRNA二级结构和 lrRNA二级结构,对郑氏比蜢线粒体基因组 srRNA和lrRNA的二级结构进行预测(Gao et al, 2009)。
2 结 果
2.1 线粒体基因组组成与基因排列
郑氏比蜢线粒体基因组序列已提交到GenBank,登录号为JF411955。本研究共测得41条可用序列,拼接后的序列总长度为 1 5618 bp, 序列覆盖度1.98倍, 除去两端重叠序列得到郑氏比蜢线粒体基因组的总长度为 1 5602 bp。它由13个蛋白质编码基因(protein-coding genes, PCGs)、22 个 tRNA 基因、2 个 rRNA 基因(lrRNA 和 srRNA)组成, 基因排列顺序(图 1)与蝗总科昆虫的排列顺序相同, 也存在tRNAAsp(D)和tRNALys(K)的倒置现象(Huang et al, 2010)。 基因排列非常紧密, 存在 14 处共 52 bp的基因重叠, 重叠区长度 1~8 bp 不等; 基因间隔序列总长度为47 bp, 共计10 处, 大小 1~20 bp之间, 基因间隔最大的在 tRNALys与 ATP8之间和ND5与 tRNAHis之间, 分别是 20 bp和10 bp; 既没有重叠, 也没有间隔的基因对共计 4 处。其中tRNAGln、tRNACys、tRNATyr、tRNAPhe、tRNAHis、tRNAPro、tRNALeu、tRNAVal和 ND1、ND4、ND4L、ND5 以及16s rRNA、12srRNA基因位于N链上, 其余23 个基因位于J 链上, 具体信息见表3。
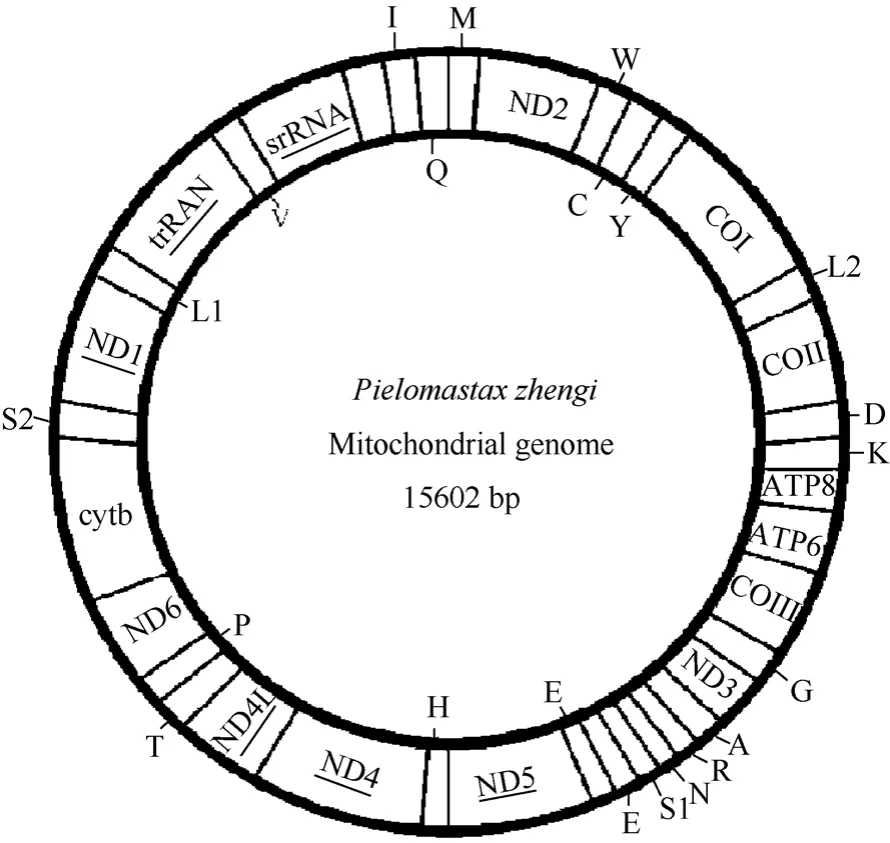
图1 郑氏比蜢线粒体基因组结构Fig. 1 The map of the Pielomastax zhengi mitogenome
2.2 线粒体基因组的碱基组成及密码子使用
郑氏比蜢线粒体基因组的碱基组成具有很强的AT 偏向性, 在4种碱基中, A含量最多, G含量最少。J链的碱基含量为: A(39.4%) > T(32.4%) >C(15.6) > G(12.6%), AT(71.8%)远大于 CG(28.2%),这种现象普遍存在于蛋白质基因、tRNA和 rRNA基因, 尤其在A+ T 富集区。郑氏比蜢的13个蛋白质基因最常用的起始密码子是 ATG, 其次是 ATC和 ATT。除COⅢ、ND3、CytB和ND1以TAG作为终止密码子, ND5的终止密码子为不完全的T外,其 8个蛋白质基因的终止密码子都为完整的 TAA,具体信息见表 3。郑氏比蜢线粒体基因组两条链上编码的蛋白质密码子使用情况存在差异, 即 J 链编码基因偏向于使用第3位点为A的密码子, 而N链偏向于使用第3位点为U的密码子。
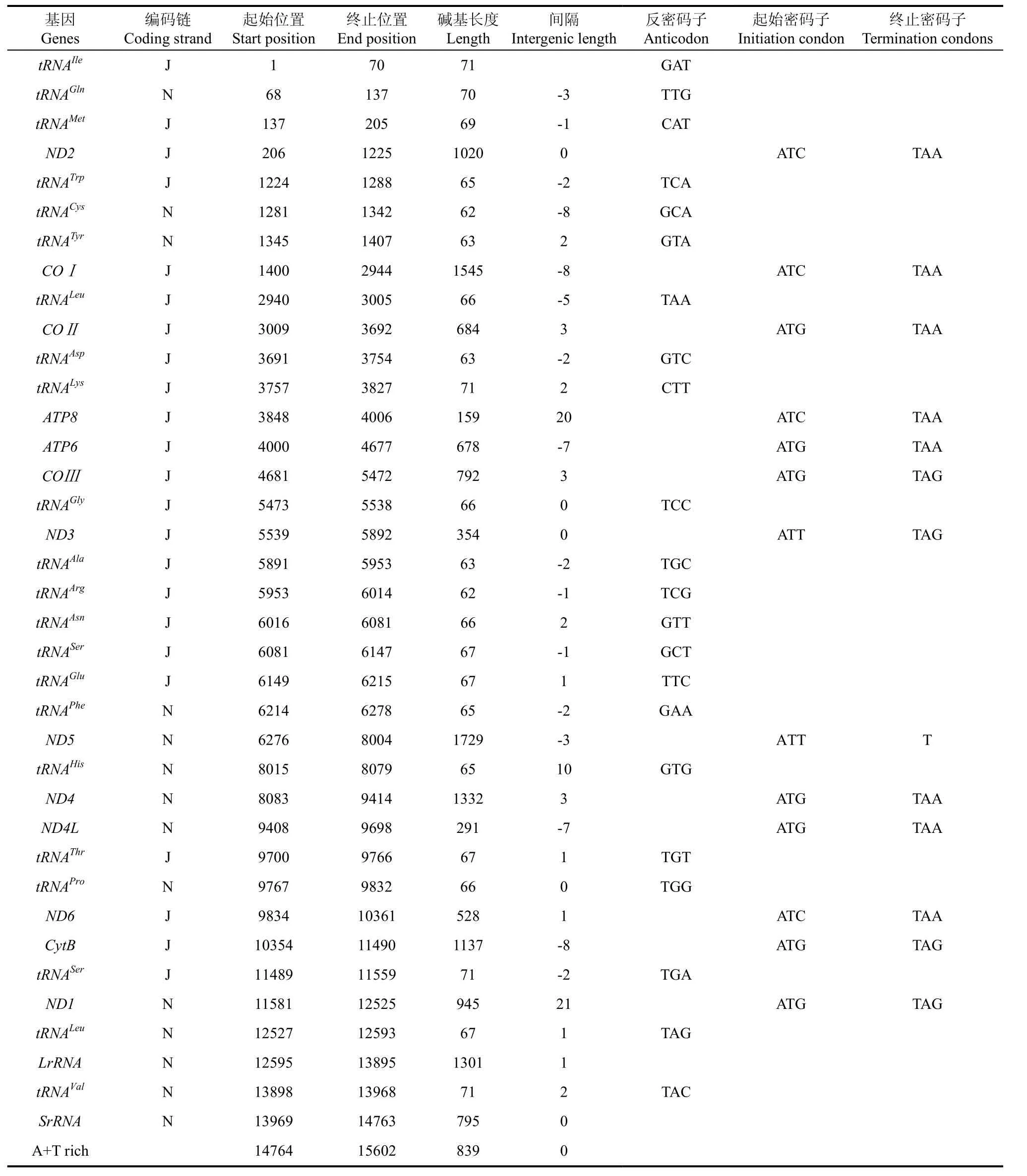
表3 郑氏比蜢线粒体基因组组成Tab. 3 Gene organization of the Pielomastax zhengi mitochondrial genome
2.3 tRNA二级结构
郑氏比蜢线粒体基因组有 22种 tRNA, 其中14种位于重链上, 8种位于轻链上。用 tRNAScan-SE 1.21 预测出郑氏比蜢16个 tRNA 的二级结构,其余 6 个 tRNA 二级结构是通过与威廉剑角蝗Acrida willemsei(NC_011303)比对确定其在整个基因组上的位置后绘出。在所预测的22 种tRNA二级结构中总共出现了37处错配, 其中有32对为GU错配; 其余 5对错配中:2对 UU 错配发生在tRNAAla和 tRNAPhe的氨基酸接受臂和反密码子臂;1对AC错配发生在tRNAMet的氨基酸接受臂; 1对AA错配发生在tRNALys的氨基酸接受臂; 1对 UC错配发生在 tRNAPro的氨基酸接受臂。除了tRNAAsp、tRNAVal和tRNASerAGN(S)的反密码环是9个碱基外, 其余的反密码环均为7个碱基(图2)。
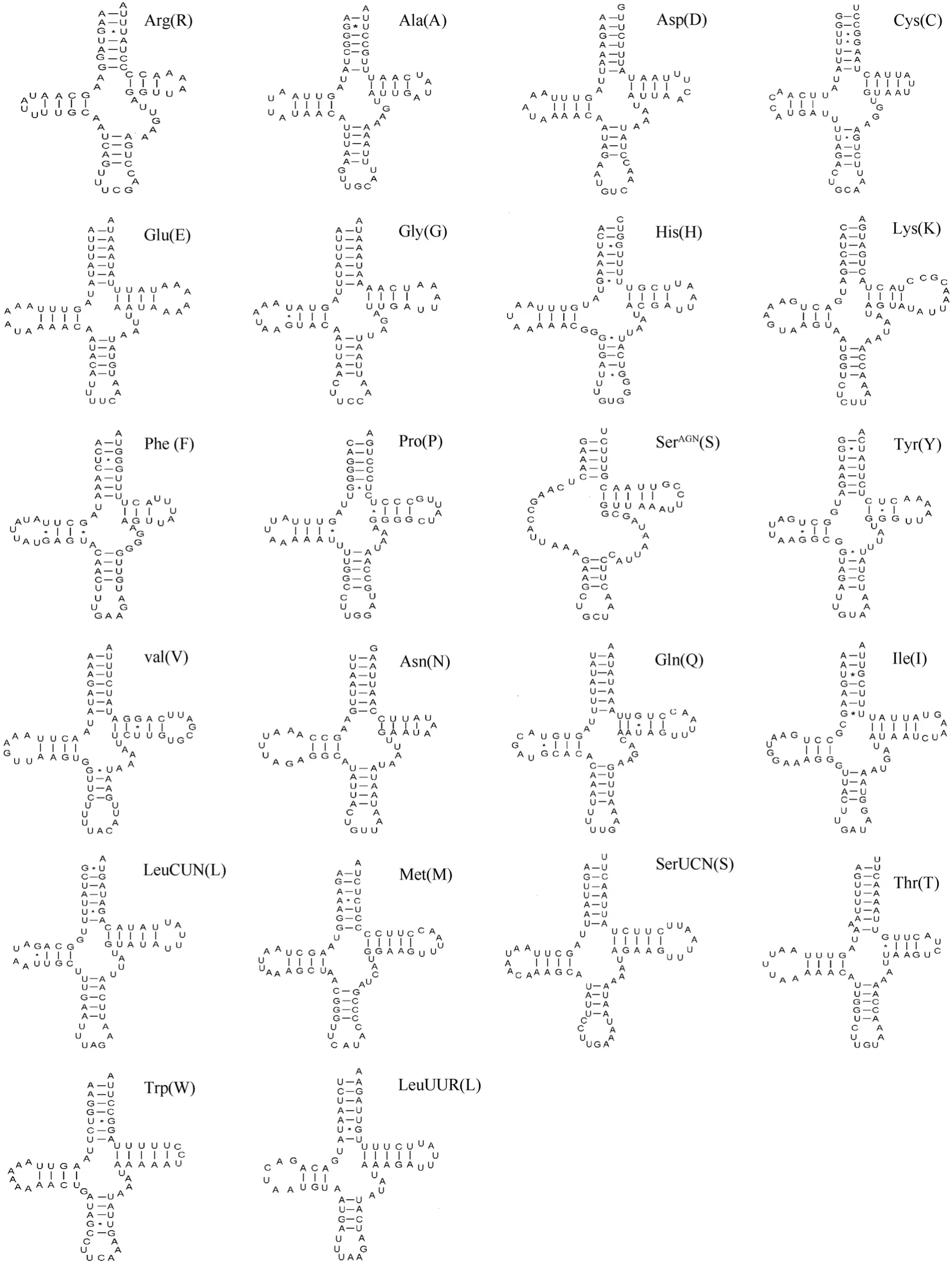
图2 郑氏比蜢线粒体基因组tRNA二级结构Fig. 2 The tRNA secondary structures of the Pielomastax zhengi mitogenome
2.4 rRNA二级结构
郑氏比蜢线粒体基因组的lrRNA和srRNA分别位于tRNALeu(L-CUN)和tRNAVal(V), 以及tRNAVal(V)和D-loop之间, 其长度分别是1 301 bp和795 bp。两个rRNA 基因的A+T含量为75.2%, 高于整个基因组的平均A+T含量(71.8%), G含量(15.1%)是C含量(9.8%)的近两倍。以短额负蝗和西藏飞蝗的lrRNA和srRNA二级结构为基础对郑氏比蜢的二级结构进行了预测。结果显示, 郑氏比蜢 lrRNA二级结构总共有6个结构域(结构域Ⅲ缺失)和44个茎环结构。srRNA的二级结构包含3个结构域和30个茎环结构(图2)。郑氏比蜢的lrRNA和srRNA二级结构与短额负蝗和西藏飞蝗基本相似, 只在少数位置存在差异。
2.5 A+T丰富区
郑氏比蜢的 A+T丰富区位于 srRNA与tRNAIle(I)之间, 全长 839 bp, A+T 含量为 82.1%,明显高于全基因组的平均水平(71.8%)。在郑氏比蜢的A+T丰富区发现一段 Poly(T)(T stretch), 且能形成一个茎环结构(图4)。
3 讨 论
3.1 基因排列顺序


图3 郑氏比蜢rRNA(lrRNA和 srRNA)二级结构预测结果Fig. 3 The secondary structures of mitochondrial rRNAs (lrRNA and srRNA)of Pielomastax zhengi
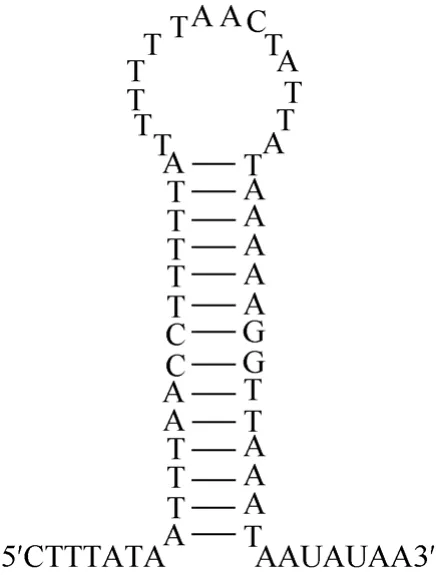
图4 A+T富集区Poly(T)及附近序列所形成的茎环结构Fig. 4 Stem-loop structure at the poly- T stretch in the A+T rich region of Pielomastax zhengi mitogenome
在直翅目昆虫线粒体基因组的研究中发现蝗亚目的蝗总科、蚤蝼总科和蚱总科基因排列次序相同, 都发生了 KD 倒置现象, 即 tRNAAsp(D)排在tRNALys(K)基因上游(DK 排列) (Ballard & Dean,2001)。蜢总科的变色乌蜢为 KD 排列, 与蝗亚目其他总科不同, 而与多数螽亚目昆虫的排序方式相同(Huang et al, 2010)。然而, 本研究所测得郑氏比蜢的基因排列顺序仍然与蝗总科的排列顺序相同,即也发生了KD 倒置现象, 形成这种排列方式的原因是由于DK这二个基因所对应的 DNA片段发生了倒置。目前关于 DNA片段倒置产生的原理, 最有说服力的是Mark和Nick的观点, 即双链断裂和重新结合模型(Zhong et al, 2005; Ye et al, 2008)。关于蜢总科昆虫的线粒体基因组排列顺序有三种可能:(1)大部分蜢总科线粒体基因组排列顺序与蝗亚目的相同; (2)大部分蜢总科线粒体基因组排列顺序与螽亚目的相同; (3)蜢总科昆虫的线粒体基因组排列顺序是随机的。蜢总科昆虫线粒体基因组排列究竟属于那种可能, 还有待研究更多蜢总科物种的线粒体基因组进一步证实, 但偏向第一种情况的可能性更大, 因为蜢总科昆虫无论从形态上分类还是从分子系统上划分都表明与蝗亚目昆虫的亲缘关系更近。
3.2 tRNA二级结构
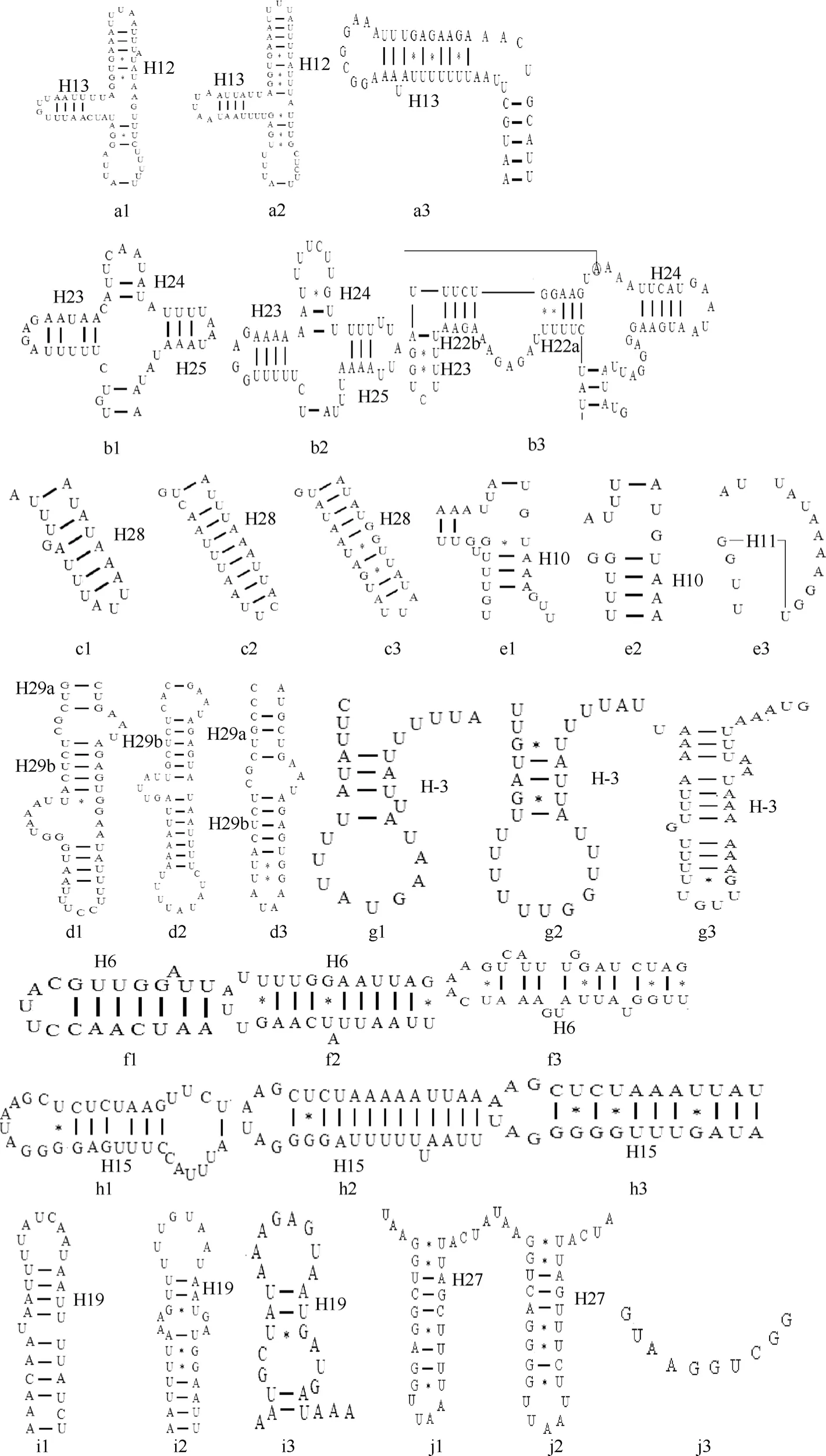
图5 郑氏比蜢、西藏飞蝗和疑钩额螽rRNA二级结构的比较Fig. 5 The comparison of rRNA secondary structures among Pielomastax zhengi, Locusta migratoria tibetensis and Ruspolia dubia
大部分直翅目昆虫线粒体基因组通过 tRNAScan-SE 1.21可以预测出20个tRNA基因。本研究的郑氏比蜢只能预测出16个tRNA基因, 除了在大部分直翅目昆虫都不能预测出来的 tRNAser(UCN)以外,还有5个tRNA 基因没有预测出来。通过比较分析发现, 除了 tRNAser(UCN)不具有典型的三叶草二级结构外, 郑氏比蜢其余的5个tRNA二级结构虽然具有典型的二级结构, 但是也存在一些差异, 即典型的三叶草结构的T¢C臂有5对配对碱基, 而这5个tRNA 二级结构的T¢C臂只有3~4对配对碱基; 另外, tRNALys和tRNAArg的反密码臂只有4对配对碱基。这些结构特征可能是导致5个tRNA基因没能预测出来的原因。另外, 预测的 22种tRNA二级结构中总共出现了 37处错配, 其中 32对为GU错配。有人认为线粒体基因组tRNA基因的部分错配可以通过 RNA编辑校正, 不会引起氨基酸转运上的障碍(Yokobori & Pääbo, 1995; Dang et al, 2008)。
3.3 rRNA二级结构
rRNA二级结构模型对于理解核苷酸的替换模式和评估 rRNA系统发育信息的可靠性非常重要(Shi et al, 2008)。预测的郑氏比蜢 lrRNA二级结构总共有 6 个结构域(结构域Ⅲ缺失)和44 个茎环结构(图2 B); srRNA的二级结构包含 3 个结构域和30 个茎环结构(图2 A)。lrRNA二级结构的结构域IV和V比较保守, srRNA二级结构的结构域I和II变异比较大。本文将郑氏比蜢的rRNA二级结构与蝗亚目西藏飞蝗的rRNA二级结构和螽亚目疑钩额螽的rRNA二级结构(Zhou et al, 2007; Jiang, 2010)进行了比较, 结果发现它们lrRNA的二级结构有四处存在明显差异(图3), srRNA的二级结构有6处存在明显差异。由此可见lrRNA要比srRNA更保守一些。另外, 通过它们rRNA二级结构的比较发现,郑氏比蜢rRNA二级结构与疑钩额螽rRNA二级结构之间的差异明显大于郑氏比蜢rRNA二级结构与西藏飞蝗rRNA二级结构之间的差异(图5)。因而,郑氏比蜢与西藏飞蝗的亲缘关系可能更近, 也就是与蝗亚目昆虫的亲缘关系更近。
3.4 A+T富集区
目前对 A+T 富集区域的研究关注主要集中于所包含的复制相关的调控信息。现在公认的是昆虫的线粒体 N链的复制起点(ON)位于控制区内, J链的复制则是在轻链的复制完成97%之后才开始, 即认为OJ(J 链的复制起点)位于距离ON97% mtDNA长度的位置上(Goddard & Wolstenholme, 1978; Saito et al, 2005)。Saito et al(2005)以果蝇为主要研究对象,同时也对10个目的20种昆虫的A+T富集区进行了研究。根据实验结果, 前期的相关研究认为, 由于ON位于控制区内, 加之其上游的一段 Poly(T)结构,它可能与复制起点的识别有关(ye et al, 2008)。本研究所分析的郑氏比蜢 A+T 富集区也有那样一段Poly(T)结构, 应该也与复制起点的识别有关。
Ballard JWO, Dean MD. 2001. The mitochondrial genome: mutation,selection and recombination[J].Curr Opin Genet Dev, 11: 667-672.
Bonfield JK, Smith KF, Staden R. 1995. A new DNA sequence assembly program [J].Nucleic Acids Res, 24: 4992-4999.
Dang JP, Liu N, Ye W, Huang Y. 2008. Complete mitochondrial genome sequence ofGastrimargus marmoratus(Thunberg) (Orthoptera :Acridoidea) [J].Acta Entomol Sin, 51(7): 671-680. [党江鹏, 刘 念,叶 伟, 黄 原. 2008. 云斑车蝗线粒体基因组全序列测定与分析.昆虫学报, 51(7): 671-680.]
Ding FM, Shi HW, Huang Y. 2007. Complete mitochondrial genome and secondary structures of lrRNA and srRNA ofAtractomorpha sinensis(Orthoptera, Pyrgomorphidae) [J].Zool Res, 28(6): 580-588. [丁方美,师红雯, 黄 原. 2007. 短额负蝗线粒体基因组及其 lrRNA 和srRNA 二级结构分析. 动物学研究, 28(6): 580-588]
Gao J, Cheng CH, Huang Y. 2009. Analysis of complete mitochondrial genome sequence ofAeropus licentiChang [J].Zool Res, 30(6):603-612. [高佳, 程春花, 黄原. 2009. 李氏大足蝗线粒体全基因组序列分析. 动物学研究, 30(6):603-612].
Goddard JM, Wolstenholme DR. 1978. Origin and direction of replication in mitochondrial DNA molecules fromDrosophila melanogaster[J].Proc Natl Acad Sci USA, 75(8): 3886-3890.
Huang Y, Liu N, Lu HM, 2010. Research progress in mitochondrial genomes of the Orthoptera insects [J].Acta Entomol Sin, 53(5) :581-586. [黄原, 刘念, 卢慧甍. 直翅目昆虫线粒体基因组研究进展.昆虫学报, 53(5) : 581-586.]
Huang JH, Huang Y, Zhou SY. 2009. Check list of Chinese species of superfamily Eumastacoidea (Orthoptera: Caelifera) [J].J Guangxi Normal Univ, 27(1): 84-87. [黄建华, 黄 原, 周善义. 2009. 中国蜢总科昆虫名录. 广西师范大学学报, 27(1): 84-87.]
Jiang DY. 2010. Determination and analysis of four complete mitochondrial genome sequences of locust populations [D]. College of Life Sciences,Xi’an Shaanxi Normal University. [江东洋. 2010. 中国四种飞蝗线粒体基因组序列的测定与分析 [D]. 西安: 陕西师范大学生命科学学院.]
Liu N, Hu J, Huang Y. 2006. Amplification of grasshoppers complete mitochondrial genomes using long PCR [J].Chn J Zool, 41: 61-65.[刘念, 胡 靖, 黄 原. 2006. 应用长 PCR 扩增蝗虫线粒体全基因组.动物学杂志, 41: 61-65.]
Saito S, Tamura K, Aotsuka T. 2005. Replication origin of mitochondrial DNA in insects[J].Genetics, 171(4): 1695-1705.
Shi HW, Ding FM, Huang Y. 2008. Complete sequencing and analysis of mtDNA inPhlaeoba albonemaZheng [J].Chn J Biochem Mol Biol, 24(7): 604-611.
Simon C, Buckley TR, Frati F, Stewart JB, Beckenbach AT. 2006.Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA [J].Annu Rev Ecol Evol Syst, 37:545–579.Wolstenholme DR, 1992. Animal mitochondrial DNA: structure and evolution [J].Int Rev Cytol, 141: 173-216.
Yokobori S, Pääbo S. 1995. Transfer RNA editing in land snail mitochondria [J].Proc Natl Acad. Sci USA, 92: 10432-10435.
Ye W, Dang JP, Xie LD, Huang Y. 2008. Complete mitochondrial genome ofTeleogryllus emma(Orthoptera: Gryllidae) with a new gene order in Orthoptera [J].Zool Res, 29(3): 236-244. [叶 伟, 党江鹏, 谢令德,黄 原. 2008. 黄脸油葫芦线粒体基因组:一种新的基因排列方式.动 物 学 研 究, 29(3):236-244.]
Zhong J, Li G, Liu ZQ, Li QW, Wang YQ. 2005. Gene rearrangement of mitochondrial genome in the vertebrate [J].Acta GenetSin, 32(3):322-330. [钟 婧, 李 光, 刘忠权, 李庆伟, 王义权. 2005. 脊椎动物线粒体DNA 的基因重排. 遗传学报, 32 (3): 322-330.]
Zhou ZJ, Huang Y, Shi FM. 2007. The mitochondrial genome ofRuspolia dubia(Orthoptera: Conocephalidae) contains a short A+T-rich region of 70 bp in length [J].Genome, 50: 855-866.
Analysis of the complete mitochondrial genome sequence ofPielomastax zhengi
YANG Hui, HUANG Yuan∗
(School of Life Sciences,Shaanxi Normal University,Xi’an710062,China)
The complete mitochondrial genome sequence ofPielomastax zhengiwas determined by using long PCR and conserved primer walking approaches. The results showed that the entire mitochondrial genome ofPielomastax zhengiis 15 602 bp long with A+T content of 71.8%. All 37 genes are conserved in the positions observed in those ofLocusta migratoria. All the genes are closely assembled by leaving 10 intergenic spacers in between. Those intergenic spacers are 47 bp in total (excluding the A+T rich region), with individual size ranges from 1 bp to 20 bp. In addition,there are a total of 52 bp overlapping sequences among 14 genes, ranging from 1−8 bp. All protein-coding genes start with a typical initiation codon in insects, ATN. Twelve protein-coding genes use the usual TAA and TAG termination codons, whereas, the ND5 genes have an incomplete termination codon (T). Except the tRNASer (AGN), whose DHU arm is absent; all the other 21 tRNA genes have typical clover-leaf secondary structures. But inPielomastax zhengi, the secondary structures of five tRNA (tRNACys, tRNALys, tRNAPhe, tRNAPro, tRNAArg) genes can not be predicted by the conventional methods as they have only 3−4, rather than 5 base pairs in the TψC arm, while, the tRNALysand tRNAArghave only 4 base pairs in the anticode arm. The predicted secondary structures of lrRNA and srRNA have 6 domains with 44 helices and 3 domains with 30 helices, respectively. The results of the rRNA secondary structure comparison showed thatPielomastax zhengiis more closely related withLocusta migratoria tibetensisthanRuspolia dubia. Like most insects, the mitochondrial genome ofPielomastax zhengihas a non-coding A+T rich region containing a polythymidine stretch, which may be involved in the replication and/or translation initiation of other genes.
Eumastacoidea;Pielomastax zhengi; Mitochondrial genome; RNA secondary structure
Q969.26; Q754
A
0254-5853-(2011)04-0353-10
10.3724/SP.J.1141.2011.04353
2011-01-06;接受日期:2011-03-28
自然基金资助项目(30670279;30970346)
∗通讯作者(Corresponding author),E-mail: yuanh@snnu.edu.cn
杨辉,硕士研究生,研究方向:基因组学
猜你喜欢
杂志排行

Zoological Research的其它文章
- Metabolism and thermoregulation between Mrs Hume’s Pheasant (Syrmaticus humiae) and Elliot’s Pheasant (S. ellioti)
- Behavioral migration diversity of the Yangtze River Japanese Eel, Anguilla japonica, based on otolith Sr/Ca ratios
- Visual modeling reveals cryptic aspect in egg mimicry of Himalayan Cuckoo (Cuculus saturatus) on its host Blyth’s Leaf Warbler (Phylloscopus reguloides)
- Afferent and efferent pathways in the visual system of the freshwater snail Planorbarius corneus
- Notch signaling dependent differentiation of cholangiocyte-like cells from rhesus monkey embryonic stem cells
- 一种有效区分移植细胞和宿主细胞脑损伤模型的建立