伴发神经系统疾病的听神经病36例临床分析
2011-01-23王锦玲王剑石力薛飞吴保仁高磊谢娟韩丽萍
王锦玲 王剑 石力 薛飞 吴保仁 高磊谢娟 韩丽萍
Starr等(1996)命名的听神经病(auditory neuropathy,AN)是由于第Ⅷ颅神经的听支病变引起的一组临床表现特殊的感音神经性聋。Starr[1]报道的10例AN患者中,8例随着病情发展出现其他周围神经症状。临床上报道的AN病例,大多为单发性,仅具有AN的听力学特征,无其他神经系统异常[2],这类AN,被称为非综合征型AN。少数AN可伴发于神经系统疾病,由于病因不同,临床表现多样,为一组症侯群,被称为综合征型AN[3]。有关神经系统疾病伴发AN的报道不多。本文分析36例神经系统疾病合并AN(综合征型AN)的临床特征及AN与神经系统疾病间的关系,并报告10例典型病例。
1 资料与方法
1.1临床资料 2001年1月至2007年9月第四军医大学西京医院耳鼻咽喉科门诊确诊的符合纳入诊断标准[4]的AN患者376例,其中AN伴发神经系统疾病者36例,男19例,女17例。年龄70天至54岁,平均20.8±11.3 岁,发病年龄平均12岁。病程70天至11年,平均3.5年。其神经系统疾病包括:弗雷德赖希(Friedreich) 共济失调5例,腓骨肌萎缩症(charcot-marie-tooth,CMT病)4例,植烷酸储积病(Refsum病)1例,视神经萎缩9例,下肢周围神经病变9例,多发性硬化2例,慢性炎症性脱髓鞘性多发性神经病(CIDP,慢性格林-巴利综合征)1例,脊髓亚急性联合变性2例,运动神经元病1例,缺血缺氧性脑病1例,红斑性肢痛症1例。患者大多来自农村,部分来自山区。
1.2方法 详细调查患者病史,36例均行纯音测听、声导抗、畸变产物耳声发射(DPOAE)、听性脑干反应(ABR)、言语识别率、眼震电图(ENG,2005年以前部分检查)、前庭诱发肌源性电位(VEMP,2005年后与ENG均常规检查),方法同前[4,5]。神经内科行全面神经系统检查,并行肌电图、运动及感觉神经传导速度(MCV、SCV)测定、腰椎穿刺检查脑脊液等;眼科行相关检查及视诱发电位;影像学检查包括:颞骨薄层CT /头颅CT、MRI。
2 结果
2.1临床表现 36例中14例(38.89%)首先出现双下肢无力、麻木、走路不稳、行走困难或视力减退,先于听力障碍1月至13年,平均1年8个月;另22例(61.11%)首先出现听不清说话,特别在嘈杂环境中,20例伴耳鸣,先于其他神经症状2月至10年,平均2年7个月。神经系统疾病的主要症状为: 下肢麻木、乏力各9例,跛行、行走困难8例,走路不稳7例,视力减退14例,头晕4例。有行走困难及耳聋家族史5例、视力减退家族史2例、新生儿黄疸史1例。
2.2纯音听阈测试 除2例小儿外,34例(68耳)纯音听力图呈低频上升型37耳(54.41%,37/68),覆盆型12耳(17.65%,12/68),平坦型12耳(17.65%,12/68),尖峰型3耳(4.41%,3/68),盆型4耳(5.88%,4/68)。根据听力图呈低频上升型或平坦型、下降型,按0.125~1 kHz或0.5~4 kHz的平均气导纯音听阈行听力损失程度的分级标准[2],轻度听力障碍4耳(5.88%),中度17耳(25.0%),中重度19耳(27.94%),重度20耳(29.41%),极重度8耳(11.76%)。
2.3言语测听 36例患者中18例(36耳)行言语识别率测试,言语识别率为61%~75%(中度障碍)4耳(11.11%,4/36),为51%~60%(重度障碍)3耳(8.33%,3/36),低于50%(极差)29耳(80.56%,29/36,其中10耳言语识别率为0)。显示言语识别率明显差于纯音听阈。
2.4声导抗测试 36例(72耳)患者行声导抗测试,其中鼓室导抗图2耳呈“AS”型,1耳呈“C1”型,其余均为“A”型曲线;静态声顺值0.2~5.3 ml。镫骨肌声反射42耳(58.3%)同侧及交叉声反射均未引出,30耳(41.67%,)的单项或多项频率声反射可引出,以1 kHz引出率高,但阈值均升高。
2.5ABR测试 36例(72耳)AN患者中,ABR均自波Ⅰ起未引出(>100 dB SPL)。
2.6DPOAE测试 所有AN患者无论纯音听阈损失程度轻重,除个别频率外,DPOAE全部可引出。
2.7前庭功能检查 26例患者行ENG冷热试验,其中半规管麻痹14例,正常12例;VEMP检测13例中9例未引出,引出的4例均幅值降低,其中2例p13及n23潜伏期延长。
2.8辅助检查 16例患者行肌电图检查,其中16例胫后神经运动神经传导速度(MCV)减慢(24~30 m/s)和/或潜伏期延长(4.1~5.8 ms)。6例腓肠神经感觉神经传导速度(SCV)减慢(0~35 m/s)及/或5例波幅下降(0~0.85 μV)。14例视诱发电位测试视觉径路传导障碍,行F-VEP检查均未引出明显P波。36例均行颅脑CT或MRI检查,除2例示颅脑有脱髓鞘病灶、1例示缺血缺氧性脑病外,其他未见异常。
2.9伴发的神经系统疾病 36例伴神经系统疾病的AN患者中23例伴前庭神经病(VN)及/或视神经萎缩(OA)同时伴发其他神经系统疾病(表1)。
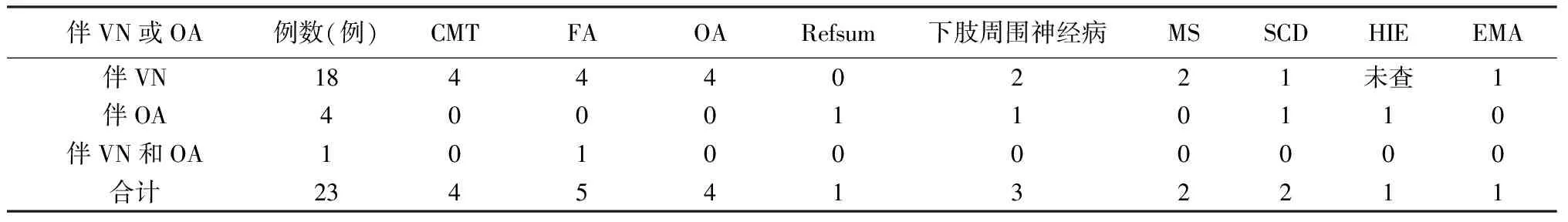
表1 23例伴前庭神经病(VN)及/或视神经萎缩(OA)AN患者同时伴其他神经系统疾病的病例数
注:CMT(腓骨肌萎缩症)、 FA(弗雷德赖希共济失调)、Refsum(植烷酸储积病)、MS(多发性硬化)、SCD(脊髓亚急性联合变性)、HIE(缺血缺氧性脑病)、EMA(红斑性肢痛症)
3 典型病例报告
3.1CMT病 病例1,女,15岁,学生,来自河南省农村。因双下肢无力、行走困难3月入住神经内科。家中三姐妹均于十二三岁时出现相同病史。神经系统检查:足下垂,呈跨阈步态,近端双下肢肌张力Ⅴ级,远端腓神经支配的肌肉肌力0级,双侧Hoffmann征(+),双侧Mayer反射、Leri反射均消失。双胫神经MCV减慢,波幅降低,双腓神经MCV减慢,波幅极低。腰椎穿刺脑脊液蛋白质增高。头颅CT、脊髓颈胸段MRI未见异常。神经内科诊断:CMT病。因双耳听力下降2年余至我科会诊,患者听不清说话,尤其在嘈杂环境中,伴耳鸣,无眩晕。纯音听力图呈中重度低频上升型,双侧镫骨肌声反射仅1 kHz于110 dB引出,DPOAE正常引出,ABR未引出,冷热试验示双侧半规管麻痹。耳鼻咽喉科诊断:听神经病,前庭神经病。住院期间予激素、营养神经等药物治疗后行走困难减轻。10年后信访,未经特殊治疗,自觉病情平稳,听力无变化,平时走路注意控制步态可与常人无异,正在外地打工。
3.2CMT病 病例2,男,7岁,来自陕西省山区。自小听力差,不会说话,3岁开始走路,走路不稳,左腿跛行。其兄6年前就诊,有相同诊断及阳性遗传病史,目前已不能行走。纯音测听双耳呈极重度聋,镫骨肌声反射消失, ABR引不出,DPOAE正常引出。冷热试验示双侧半规管麻痹,VEMP双侧未引出。耳鼻咽喉科诊断:听神经病,前庭神经病。MCV检测双侧胫神经运动传导速度减慢,左腓神经潜伏期延长,波幅明显降低,右侧偏低;SCV检测右侧腓神经感觉神经传导速度轻度减慢,提示双侧胫、腓神经受损(以左侧腓神经为显)。颅脑CT未见异常。神经内科诊断:CMT病。经用营养神经、激素及改善微循环等药物治疗,跛行短期减轻。
3.3Refsum病 病例3,男,16岁,学生,来自陕西省山区。因视力减退3年、听力减退2年、双下肢无力2月入住神经内科。神经系统检查:双下肢肌力3级,腱反射减低,双下肢肌肉萎缩,病理征阳性,共济运动障碍。肌电图示左胫、腓神经波幅偏低。肌酶检测示谷草转氨酶41 IU/L,乳酸脱氢酶(LDH)239 IU/L,磷酸肌酸激酶(CPK)2 074 IU/L,CPK/LDH 8.68。双眼视力0.01,双视乳头色苍白,视野向心性缩小,视诱发电位显示双视觉径路传导障碍。神经内科诊断:Refsum病,视神经萎缩。行纯音听阈测试呈中重度平坦型听力图,双侧镫骨肌声反射仅1 kHz于110 dB引出,DPOAE正常引出,DPOAE对侧声抑制消失,ABR未引出,ENG正常。耳鼻咽喉科诊断:听神经病。
3.4Friedreich共济失调 病例4,女,17岁,学生,来自陕西省山区。双耳渐进性听力下降、听不清言语声4月余,偶有耳鸣。半年前双下肢麻木,走路摆动不稳,闭眼身体向一侧倾倒。有家族史。神经内科检查:四肢腱反射普遍低下,肌张力降低,四肢共济失调征阳性,Romberg征及直线行走试验阳性,闭目时明显。冷热试验示双侧半规管麻痹,无视动中枢异常;重心平衡试验为弥漫型动摇型,提示重度平衡功能障碍。胫神经MCV减慢。颅脑MRI未见异常。神经内科诊断:Friedreich共济失调。纯音测听呈双耳低频上升型中度听力损失,镫骨肌声反射未引出,DPOAE正常引出,ABR未引出。耳鼻咽喉科诊断:听神经病,前庭神经病。
3.5遗传性视神经萎缩 病例5,男,15岁,双眼视力明显减退6月,听力减退4月,其兄亦有视物不清病史。双眼视力0.15,双视乳头色淡,中心视野(灰度值):左404、右414,P-VEP:双视觉径路传导障碍,P100波潜伏期左113 ms、右117 ms。颅脑CT未见异常。眼科诊断:遗传性视神经萎缩。双耳纯音听力图呈中重度低频上升型,镫骨肌声反射未引出,DPOAE正常引出,ABR未引出,冷热试验示双侧半规管麻痹。耳鼻咽喉科诊断:听神经病,前庭神经病。
3.6脊髓亚急性联合变性 病例6,女,42岁,护士,来自陕西省某县城。因进行性双下肢无力4月,视物模糊2周、双耳听力下降6月,入住神经内科。骑车及行走100米左右时即感双下肢无力,行走困难。双下肢肌力Ⅳ级,膝腱反射减弱,T8以下深感觉减弱,Romberg征(+)。双胫神经SEP P40潜伏期延长。血液IgE↑、IgG↓、C3↑,脑脊液IgG↑、IgM↑,血乳酸脱氢酶(LDH)↑。颅脑MRI示左大脑脚点状脱髓鞘。视诱发电位示视觉径路传导机能减退。神经内科诊断:脊髓亚急性联合变性,视神经萎缩。纯音听力图双侧呈中及中重度覆盆型,言语识别率右耳40%,左耳32%。双耳DPOAE可引出,ABR未引出,ENG正常。VEMPp13、n23 潜伏期左、右耳分别为28.61、37.29 ms及30.28、37.63 ms,振幅为40.77 μV及47.12 μV。耳鼻咽喉科诊断:听神经病,前庭神经病。
3.7慢性格林-巴利综合征 病例7,女,17岁,学生,来自陕西省农村。双下肢无力,抬腿困难反复发作4年余,听力减退1年。4年前曾在北京及2年前在西安某部队医院均诊断为慢性格林-巴利综合征。腱反射减弱,胫后神经MCV潜伏期延长,腓神经SCV波幅降低。CSF蛋白增高。颅脑MRI(-)。纯音听力图呈中度尖峰型,言语识别率双耳40%,DPOAE可引出,ABR未引出,ENG正常。耳鼻咽喉科诊断:听神经病。
3.8多发性硬化 病例8,女,35岁,来自陕西省山区。因双下肢麻木、行走不稳1年半3次入住神经内科,听力减退2年。Romberg征阳性,双侧肱二、三头肌腱反射(++),直线行走试验(+),右胫前神经MCV未引出电位,左胫前神经MCV波幅降低,双胫神经P40波幅潜伏期延长。CSF蛋白0.4 g/L,CSF IgG 41 mg/L,IgA 3 mg / L,IgM 1.14 mg/L。MRI示侧脑室前角局部有脱髓鞘病灶。神经内科诊断:多发性硬化。纯音听力图呈双耳低频上升型中重度听力损失,言语识别率双耳0%,DPOAE可引出,ABR未引出。冷热试验示双侧半规管麻痹,VEMP双侧未引出。耳鼻咽喉科诊断:听神经病,前庭神经病。
3.9运动神经元病 病例9,女,54岁,工人,来自西安市。因四肢无力2个月入住神经内科。半年前感冒后出现听力减退、耳鸣,2月前感冒后出现双下肢无力、声音嘶哑,继而双上肢无力,行走费力。双侧肱二、三头肌腱反射(++),双跟膝腱反射(+++),肌电图呈神经元性改变,主动收缩时运动单位时限增加,双胫前神经波幅降低。颅脑MRI(-)。神经内科诊断:运动神经元病。电子喉镜检查显示双声带运动好,闭合稍欠佳。纯音测听示双耳尖峰型中重度听力损失,DPOAE可引出,ABR未引出。耳鼻咽喉科诊断:听神经病。
3.10下肢周围神经病 病例10,男,14岁,学生,来自陕西省农村。听力减退2.5年,双下肢麻木5个月。跟膝腱反射稍降低,胫前神经MCV:左23 m/s,右28 m/s,潜伏期右4.6 ms,左4.9 ms;腓肠神经SNV波幅0.5 μV,传导速度正常。颅脑MRI(-)。神经内科诊断双下肢周围神经病。纯音测听示双耳低频上升型中度听力损失。言语识别率双耳40%,DPOAE可引出,ABR未引出。冷热试验示双侧半规管麻痹,VEMP双侧未引出。耳鼻咽喉科诊断:听神经病,前庭神经病。
4 讨论
本组综合征型AN的临床特点主要表现为下肢无力,麻木,行走困难,或严重视力下降,并伴听力减退,听不清说话,尤其在嘈杂环境中,但均具有典型的AN听力学特征。从发病病程看,22例(61.11%) 患者首先出现听力障碍,病程2月至10年(平均2年7个月)后出现其他神经症状,这些患者多先就诊于耳鼻咽喉科,仅14例(38.89%)患者首先出现下肢麻木、无力、行走困难或视力下降,1月至13年(平均1年8个月)后出现听力障碍,但这些患者多先就诊于神经内科或眼科。本组AN的听力学检测结果与非综合征型AN的特征相同。26例行前庭功能ENG冷热试验、VEMP检测,19例显示前庭功能障碍而诊断为前庭神经病,其发病率较高,可能与耳蜗神经和前庭神经解剖关系密切、耳蜗神经病变波及前庭神经有关。关于AN与前庭上、下神经的检测及关系已有报道[4]。在神经电生理检查中,视神经萎缩患者经眼科检查及视诱发电位检查可呈视觉径路传导障碍。下肢周围神经病变患者经运动及感觉神经传导速度(MCV、SCV)及肌电图检测,可见传导速度减慢、潜伏期延长及波幅降低,这是一有助于评定周围运动及感觉神经传导功能的诊断技术。本组颅脑CT或MRI除2例示颅脑有脱髓鞘病灶、1例示缺血缺氧性脑病外,其他未见异常。
本组综合征多为神经系统遗传性周围性疾病,目前其病因及发病机理已部分阐明。本组中4例AN伴发于腓骨肌萎缩症,其中2例有明确家族遗传史,属于遗传性运动感觉神经病(HMSN)范畴,分I型脱髓鞘型(HMSN-I)和Ⅱ型轴索变性型(HMSN-Ⅱ)。脱髓鞘型多为常染色体显性遗传、少数为常染色体隐性遗传及X-连锁遗传方式。致病基因定位于17p11.2~p12,该基因编码周围神经髓鞘蛋白22 (PMP22),它的重复突变导致PMP22过度表达,使周围髓鞘蛋白增加。以足内侧肌和腓骨肌萎缩弛缓性肌无力、弓形足、运动神经传导速度减慢及轻微感觉障碍为特征,部分病例可伴有耳聋、视神经萎缩及共济失调等症状[6]。2000年Leonardis[7]对一吉卜赛家族4代19名个体的研究发现,3名确诊为HMSN患者均伴有听力损失,且均符合AN的特征。Guergueltcheva 2006年[8]检查了两例常染色体隐性遗传的CMT患者,发现除有运动感觉神经障碍外,并有AN的听力学特征。本组病例1患病10年后随访,病情稳定,并能外出打工,表明预后尚好,对症处理可提高患者生活质量。
本组5例Friedreich共济失调,又称少年型或脊髓型共济失调,为常染色体隐性遗传,由位于9号长臂(9q12.1~q13)基因缺陷所致。平均13岁前后发病,是一组临床表现以共济失调为主的神经系统遗传变性病,主要发生于脊髓,也可发生于小脑、脑干及大脑,可伴其他系统异常,如前庭及听力障碍、视力减退及视神经萎缩等。有报道其听力损失符合听神经病的特征[9]。本组5例均伴听神经病及前庭神经病,其中1例同时伴视神经萎缩。
本组1例Refsum病(最近归类为HMSN-Ⅳ型)伴有听神经病及视神经萎缩。患者因双下肢无力、视物不清、听不清就诊于神经内科。Refsum病又名植烷酸储积病,为常染色体隐性遗传病。其三大主要临床特征为:视网膜变性(夜盲)、多发性周围神经病、小脑性共济失调。病因是由于α-羟化酶缺乏,体内植烷酸不能代谢而沉积于脂质所致[10]。Weiller等[11]报道3例HMSN-Ⅳ型伴视神经萎缩及感音神经性聋,电生理检测符合听神经病。
视神经萎缩是由各种相关疾病导致视网膜神经节细胞及其轴突变性、萎缩的一种病理改变。有报道,由于线粒体功能缺陷引起的遗传性视神经病中以常染色体显性遗传性视神经萎缩(DOA或ADOA)即Kjer型最常见,Leber遗传性视神经萎缩(LHON)较少见[12]。Huang等[13]提出常染色体显性遗传视神经萎缩可伴有听力损失,并符合AN的听力学特征,其突变位点在 OPA1 基因的R445H,发病机制可能为听神经末梢的脱髓鞘改变所致,人工耳蜗植入术后的电刺激有效。Amati-Bonneau等[14]发现5名合并视神经萎缩和耳聋的患者出现OPA1基因R445H突变的杂合子,听力检测均符合AN;其皮肤成纤维细胞显示线粒体网状系统为超碎片状,线粒体膜电位降低,三磷酸腺苷合成缺陷;此外,OPA1在耳蜗感觉神经细胞中广泛表达。因而提出视神经萎缩和耳聋可能与线粒体网状系统为超碎片状引起的能量缺乏有关。Mizutaxi[15]报道一日本OPA1基因突变患者伴听神经病及前庭功能障碍,冷热试验未引出眼震,VEMP左耳未引出。Leber遗传性视神经萎缩是由于线粒体mtDNA基因突变主要累及视网膜、巩膜筛板前部视盘黄斑束纤维从而导致视神经退行性变的母系遗传病。G11778A、G3460A、T14484C是目前公认的致病性最强的三种原发性突变位点。Yu-Wai-Man等检测10例LHON基因突变患者的听力,未发现听神经病征象[16]。本组9例视神经萎缩均伴听神经病,伴前庭神经病4例,伴发于Friedreich共济失调、Refsum病、下肢周围神经病、脊髓亚急性联合变性及缺血缺氧性脑病各1例。根据临床表现如视力明显减退、视乳头色淡或苍白、视野向心性缩小及视诱发电位显示视觉径路传导障碍,波幅降低,潜伏期延长或引不出等临床可作出诊断。本组2例视神经萎缩有明确阳性家族史,2例伴发于遗传性病Friedreich共济失调及Refsum病,根据其临床表现可提示为DOA。有条件者应行基因检测以明确其遗传属性。
多发性硬化是以中枢神经系统多灶炎症及脱髓鞘病变为特点的自身免疫病,可能是遗传易感个体与环境因素作用而发生的自身免疫过程。有报道多发性硬化患者伴听力损失者约为3%,ABR波Ⅰ异常率为2%`~10%。本组2例多发性硬化伴听力障碍,听力学检测呈AN的听力学特征,ABR波Ⅰ消失,表明中枢性脱髓鞘病变亦可能累及听神经。慢性格林-巴利综合征是周围神经的慢性复发性炎症性脱髓鞘性多发性神经病的自身免疫过程,本组1例曾经北京及西安两部队医院予以诊断,其伴听力减退,听力学检测符合AN的诊断,提示AN发病可能与自身免疫过程有关。
本组部分伴发AN的神经系统变性病病因不明。2例脊髓亚急性联合变性中,一例伴有前庭神经病,一例伴有视神经萎缩,病因可能由于维生素B12缺乏引起的神经系统变性周围神经疾病。1例54岁的运动神经元病患者,发病前有感冒史,肌电图呈神经元变性改变,是一组选择性侵犯脊髓前角细胞的慢性进行性变性疾病,病因可能与病毒感染、免疫、中毒因素有关。1例红斑性肢痛症,可能由于周围性自主神经功能障碍,使末梢血管极度扩张所致,患者伴有听神经病及前庭神经病。1例仅出生70天的新生儿,系7个半月早产,有新生儿黄疸史,外院诊断为缺血缺氧性脑病,患儿DPOAE筛查通过,但AABR(自动ABR)引不出,并伴视神经萎缩,提示患儿高胆红素血症、缺血缺氧不仅损害脑部中枢,也损害听神经及视神经。本组下肢周围神经损害9例,病因不明,患者除伴发AN外,仅表现下肢无力麻木,尚无其他神经症状,有称之为下肢单神经病,是否病变尚处于早期阶段,随着病情发展而出现其他神经损害,有待随访观察。
分析本组神经系统疾病,特别是遗传性、周围性神经病的病理基础多为神经脱髓鞘病变,如CMTⅠ型为脱髓鞘型,病理表现为神经纤维呈对称性节段性脱髓鞘,部分髓鞘再生,雪旺细胞增生与修复,形成“洋葱头”样结构,造成运动和感觉神经传导速度减慢[6]。Friedreich共济失调症为脊髓后索、脊髓小脑束和皮质脊髓束变性,周围神经脱髓鞘,胶质增生,而脑干、小脑和大脑受累较轻[6]。Refsum病患者除器官有大量脂质储积外,还有周围神经增厚,髓鞘广泛脱失,有洋葱样改变[6]。本组CIDP 1例,CIDP是周围神经的慢性复发性疾病,病理以脱髓鞘与髓鞘再生并存,出现“洋葱头样”改变[6]。至于AN相关部位的病理学改变报道很少。根据听力学检测分析,多认为听神经耳蜗支纤维可能存在不均匀节段性脱髓鞘病变,内毛细胞与Ⅰ型螺旋神经节细胞突触连接间的病变,使内毛细胞合成和释放神经递质的非同步化,导致神经冲动排放的非同步化,这可解释言语分辨率低、DPOAE正常而ABR异常。梁志坚对57例多发性硬化患者行ABR检测,发现部分患者存在听神经损害,表现为ABR波Ⅰ潜伏期延长或波Ⅰ消失,有的MRI还可观察到听神经颅骨内段条索状长T1 长T2 异常信号,听神经肿胀,认为听神经远端发生脱髓鞘病变是其可能的病理生理机制[17]。本组采用VEMP检测前庭下神经,13例中9例未引出,引出的4例均幅值降低,其中2例p13及n23潜伏期延长,可能为前庭下神经的脱髓鞘病变所致,这也为AN患者耳蜗神经可能存在脱髓鞘病变提供了支持。综上所述,本组神经系统疾病与伴发的AN多以脱髓鞘病变为病理基础,提示影响原发神经系统疾病的病理过程也可影响听神经。
本组患者神经系统疾病的临床诊断主要根据临床症状和多项辅助检查由神经内科作出,尚缺乏病理活检及基因诊断依据,尚需进一步从分子和基因、免疫学、病理学水平等方面,进一步阐述和揭示综合征型AN的发病机制,进而对其临床早期防治带来希望。
5 参考文献
1 Starr A, Picton TW, Sininger Y, et al. Auditory neuropathy[J]. Brain, 1996, 119: 741.
2 王锦玲,石力, 薛飞, 等. 听神经病听力学特征及病损部位分析[J]. 听力学及言语疾病杂志,2007,15:89.
3 Varga R, Kelley PM, Keats BJ, et al. Non-syndromic recessive auditory neuropathy is the result of mutations in the otoferlin (OTOF) gene[J]. J Med Genet, 2003 ,40:45.
4 王锦玲,石力,孙伟,等.听神经病伴发前庭上、下神经损害及与听神经病的关系[J].听力学及言语疾病杂志,2006,14:405.
5 王锦玲,高磊,薛飞,等. 听神经病的临床与听功能特征[J]. 临床耳鼻咽喉科杂志,2002,16:518.
6 陈嵘,王莹,侯国庆. 遗传性周围神经病[M]. 见:梁秀龄,主编.神经系统遗传性疾病. 北京:人民军医出版社,2001.62~73, 96~99,274~275.
7 Leonardis L, Zidar J, Popovic M, et al.Hereditary motor and sensory neuropathy associated with auditory neuropathy in a Gypsy family[J]. Pflugers Arch,2000,439:208.
8 Guergueltcheva V, Tournev I, Bojinova V, et al. Early clinical and electrophysiologic features of the two most common autosomal recessive forms of charcot-marie-tooth disease in the roma [J]. J Child Neurol, 2006,21:20.
9 Lopez-Diaz-de-Leon E, Silva-Rojas A. Auditory neuro-pathy in Friedreich ataxia. A report of two cases [J].Int J Pediatr Otorhinolaryngol, 2003,67:641.
10 Wierzbicki AS, Lloyd MD, Schofield CJ, et al. Refsum's disease:A peroxisomal disorder affecting phytanic acid alphaoxidation[J]. J Neurochem, 2002, 80:727.
11 Weiller C, Ferbert A. Hereditary motor and sensory neuro-pathy (HMSN) and optic atrophy (HMSN type VI, Vizioli) [J]. Eur Arch Psychiatry Clin Neurosci,1991,240:246.
12 张艳玲,刘祖国.常染色体显性视神经萎缩的研究进展[J].国际眼科纵览,2006,30:206.
13 Huang T, Santarelli R, Starr A, et al. Mutation of OPA1 gene causes deafness by affecting function of auditory nerve terminals [J]. Brain Res,2009 ,1300:97.
14 Amati-Bonneau P, Guichet A, Olichon A, et al. OPA1 R445H mutation in optic atrophy associated with sensorineural deafness [J].Ann Neurol,2005,58:958.
15 Mizutari K, Matsunaga T, Inoue Y,et al. Vestibular dysfunction in a Japanese patient with a mutation in the gene OPA1[J]. J Neurol Sci, 2010, 15:293:23.
16 Yu-Wai-Man P, Elliott C, Griffiths PG ,et al, Investigation of auditory dysfunction in Leber hereditary optic neuro-pathy[J].Acta Ophthalmol, 2008 ,86: 630.
17 梁志坚,黄建敏. 多发性硬化听神经末梢损伤伴发神经损害的研究[J].广西医学杂志,2004,26:488.
