具有自激活切割活性肠激酶轻链基因设计、表达及活性研究
2011-01-06朱文赫郭志刚刘红建郭祥瑞孙德军
朱文赫,郭志刚,刘红建,郭祥瑞,孙德军
(1.吉林大学再生医学科学研究所 生物技术药物教研室,吉林 长春 130021;2.吉林大学 中日联谊医院 骨科,吉林 长春 130033)
具有自激活切割活性肠激酶轻链基因设计、表达及活性研究
朱文赫1,郭志刚2,刘红建1,郭祥瑞1,孙德军1
(1.吉林大学再生医学科学研究所 生物技术药物教研室,吉林 长春 130021;2.吉林大学 中日联谊医院 骨科,吉林 长春 130033)
目的 获得 Asp-Asp-Asp-Asp-Lys序列 +牛 EKL的 cDNA序列,实现 EKL基因在大肠杆菌中的融合表达和自切割。方法 用 Trizol法从牛十二指肠组织中提取总 RNA,利用 RT-PCR技术扩增其 cDNA片段,将此片段克隆于表达载体 p GEX-2T,并在其前引入肠激酶 (EK)识别序列。结果 所表达蛋白经 SDS-PAGE分析,相对分子质量约为 61 000,经 GST-Sepharose亲和色谱柱纯化后得到单一蛋白,SDS-PAGE显示单一条带,用微量 EK引发自切割,切断 GST标签蛋白和 Asp-Asp-Asp-Asp-Lys序列,采用对氨基苯甲脒-Sepharose亲和色谱获得了轻链 EK的单体。结论 成功地克隆、表达了牛 EK轻链基因,采用自激活、切割获得了 EK单体,每 1 L培养基获得 EK 5 mg,为进一步进行重组牛 EK活性的研究及应用奠定了基础。
肠激酶;大肠杆菌表达系统;谷胱甘肽转移酶;自激活
肠激酶 (Enterokinase,EK)是存在于哺乳动物十二指肠内的一种异源二聚体丝氨酸蛋白酶,EK相对分子质量 (Mr)为 150 000,由 1条 Mr115 000重链和 1条Mr35 000轻链组成。重组融合蛋白经 EK酶解所释放的目的蛋白质 N末端氨基酸序列具有和野生型相同的一级结构[1],EK由于专一识别 5个氨基酸多肽 (D-D-D-D-K-X1)并在赖氨酸的羧基上切割,是 N末端融合通常选择的蛋白酶,在其他残基的不规则切割发生水平较低,这取决于蛋白质底物的构象。1个单位定义为在 23℃下切割融合蛋白 50μg在 8 h内达到 95%切割率所需的酶量,这种特点使得 EK成为基因工程融合蛋白表达后修饰过程中 1个极其有用的工具而被广泛应用。天然EK由 1条结构亚基 (重链)和 1条催化弧基 (轻链)构成[2],两者通过 1个分子间二硫键结合,结构亚基负责将催化亚基固定在小肠刷状缘膜上并引导它向肠腔移动,催化亚基可以特异性识别 A sp-Asp-A sp-Asp-Lys序列并沿序列的羧基端切下[3]。EK轻链结构在人、牛和猪中相对保守,其识别序列 Asp-A sp-Asp-Asp-Lys在脊椎动物中也有很强的保守性,且几乎所有被定序的胰蛋白酶原都具有 4个天冬酰胺相连的特征,此序列在其他的天然蛋白质上又非常罕见[4],而 EK的活性中心有 1个特殊的阳离子位点,使得有强大负电的 Asp-Asp-Asp-Asp可以与此阳离子位点结合。EK轻链体外具有全酶酶切特异性,相对于天然 EK,表现出对基因工程融合蛋白质底物的酶切活性增强[5]。
天然的 EK来源非常有限,目前获得 EK的主要方法是从猪、牛等动物的十二指肠中分离提取得到,操作步骤繁琐,提取分离的成本高,而且天然提取的EK易为其它蛋白酶污染,在切割融合蛋白质的同时又降解了目的产物[6],大大影响了其商品价值。本研究中,我们设计并构建了 p GEX-EKL原核融合表达载体,为经济、有效切除 GST部分并获得与天然结构一致的 EK,在 EK序列的 N端引入 EK位点,实现 EK的切割后的自激活和切割,将其在大肠杆菌中表达,并对表达产物进行纯化、鉴定,为进一步进行重组牛 EK活性的研究及应用奠定了基础。
1 材料和方法
1.1 材 料
1.1.1 菌株和质粒 牛十二指肠购于长春皓月集团 ;菌株 E.coli DH 5α(DE3)、BL21(DE3)和 p GEX-2T质粒由吉林大学生物药物研究室保存;pMD18-T载体购于大连宝生物工程有限公司。
1.1.2 主要试剂 所有的限制性内切酶、T4酶、Taq DNA聚合酶、Turb RT反转录酶、蛋白质Marker、IPTG均购于大连宝生物工程有限公司;Trizol购于北京鼎国生物技术有限责任公司;蛋白胨、酵母提取物、琼脂糖均购于 Oxiod公司。
1.2 方 法
1.2.1 EKL基因的获得 牛十二指肠组织置于预冷的组织研磨器中研磨使其匀浆化,用 Trizol法抽提总 RNA,用反转录试剂盒 RT-PCR得到相应的cDNA。根据 GenBank上牛 EK的全基因序列设计PCR引物,为了便于表达载体的构建,引入了 Bam HⅠ和 Eco RⅠ的酶切位点:T1(5′-CGGGATCCCCACCATGGAUGAUGAUGAUAAGATTGTCGGAGGAAG)和T2(3′-CCGGAATTCTACATCTTTTGAAACATAGGTGAGAC)。产物经琼脂糖凝胶电泳鉴定正确后用 Agarose Gel DNA Fragment Recovery Kit(TaKaRa)胶回收试剂盒回收。
1.2.2 EKL原核表达质粒的构建 将 PCR获得的EKL基因片段连接到 pMD18-T载体上并转化E.coli BL21,涂布于含有氨苄青霉素 (Amp)的琼脂板上,挑取阳性克隆并进行菌落 PCR鉴定和酶切鉴定。将经过 E.coli BL21克隆扩增的 pMD18-EKL和原核表达载体 p GEX-2T用 Bam HⅠ、Eco RⅠ双酶切,产物回收后用 T4-DNA连接酶连接,连接产物转化E.coli DH 5α感受态细胞,用 Bam HⅠ、Eco RⅠ双酶切鉴定质粒,酶切正确的质粒经由上海生工生物工程技术服务有限公司进行测序。
1.2.3 融合蛋白质 GST-EKL的诱导表达 在p GEX-2T表达系统中融合蛋白质的表达受Lac启动子的调控,需要 IPTG进行诱导。挑选转化了 p GEX-rEKL的单克隆接种于含 Amp(100μg/mL)的 2×YT液体培养基 100 mL中,200 r/min 37℃培养至A600为 0.8左右,加入 IPTG至终浓度 0.8 mmol/L,在 25℃培养 3 h。
1.2.4 融合蛋白质 GST-EKL的纯化 将诱导表达的重组菌 4℃,6 000 r/min离心 20 min收集菌体,按每 1 mL培养物用去离子水 25μL重悬菌体,加入溶菌酶至终浓度 100μg/mL,室温放置 15 min。然后按每 1 mL培养物加入预冷的 pH 7.4,0.05 mol/L磷酸盐缓冲液 (PBS)25μL,按破碎体积选择合适的超声波探头,进行超声破碎。破碎后加入 20%Triton X-100至终浓度为 1%,混匀,放置 30 min,12 000 r/min离心 10 min,获得超声破碎的上清和包涵体沉淀。将包涵体依次置于洗涤缓冲液 (0.02 mol/L Tris-HCl,0.15 mol/L NaCl,1%Triton X-100,pH 7.5)、破菌缓冲液 (0.02 mol/L Tris-HCl,0.15 mol/L NaCl,pH 7.5)、变性缓冲液 (0.02 mol/L Tris-HCl,0.15 mol/L NaCl,8 mol/L尿素,0.1 mol/Lβ-巯基乙醇,pH 7.5)中进行溶解,4℃透析至尿素终浓度为 0.1 mol/L,离心后上清液与超声破碎后的上清合并后与谷胱甘肽 Sepharose 4B柱结合,依次用PBS和谷胱甘肽洗脱液洗 3次,收集洗液进行 SDSPAGE分析。
1.2.5 融合蛋白质 GST-EKL的酶切 将含有融合蛋白质的谷胱甘肽洗脱液透析除盐后,进行 EK切割,按每 100 mg融合蛋白质加 EK 0.1μg,23℃酶切过夜,然后取样进行 SDS-PAGE检测,反应后的体系通过对氨基苯甲脒-Sepharose 4B凝胶柱,含 0.5 mol/L NaCl的 Tris-HCl缓冲液 (pH 8.0)洗脱 ,收集洗脱峰,即为重组 EK。
1.2.6 重组 EK的活性检测 将纯化后的重组 EK对重组融合蛋白质 GST-金黄色葡萄球菌蛋白 A进行酶切,重组 EK与融合蛋白质的比例分 1∶100,23℃酶切过夜,然后取样进行 SDS-PAGE检测。
2 结 果
2.1 EKL DNA片段的 PCR扩增及原核表达质粒的构建
用 RT-PCR扩增获得大小约 700 bp的目的片段 (图 1),将片段纯化酶切后克隆于经同样酶切的p GEX-2T载体上,用 Bam HⅠ、Eco RⅠ双酶切鉴定,片段大小正确 (图 2)。
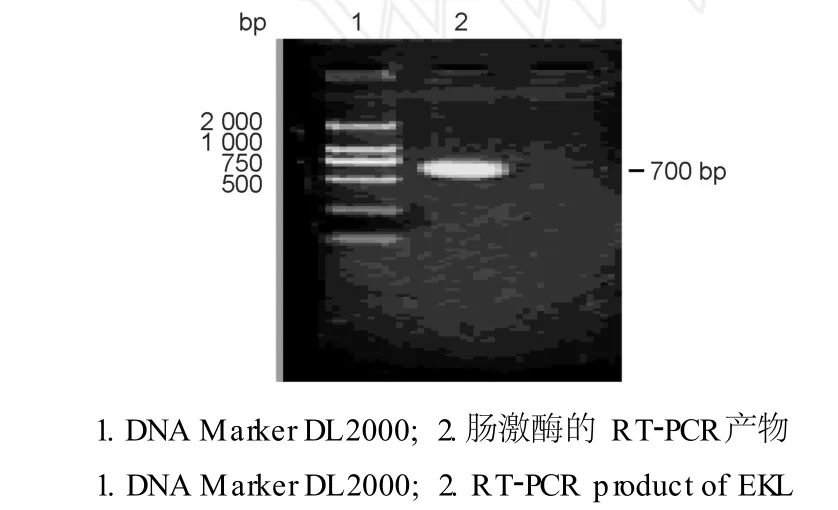
图1 EKL DNA片段的 RT-PCR扩增Fig.1 The amplified EKL fragment by RT-PCR
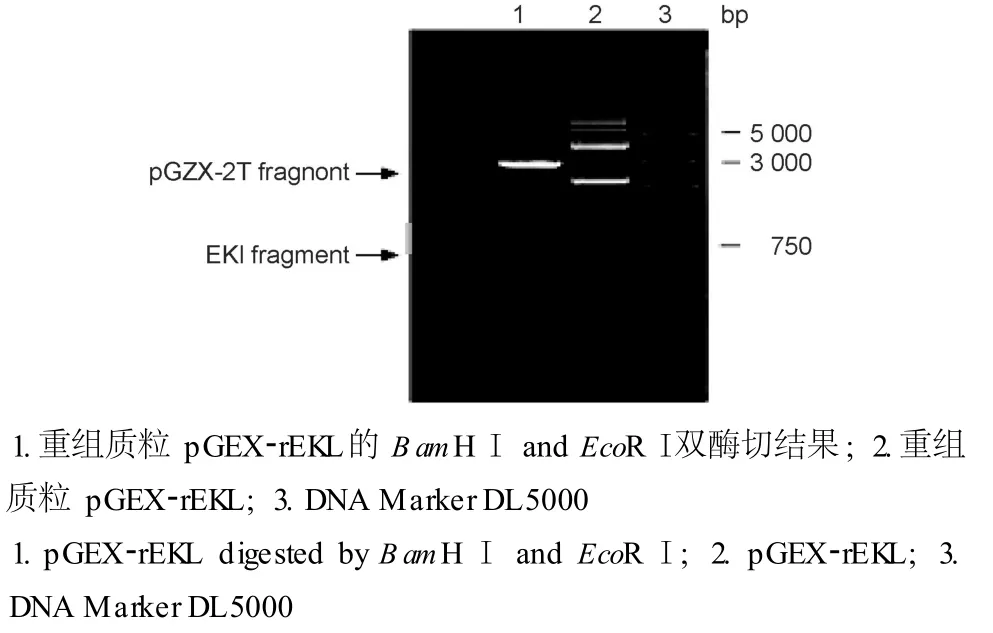
图2 重组质粒的酶切鉴定Fig.2 Identification of the recombinant plasmid by digestion with Bam HⅠ and Eco RⅠ
2.2 融合蛋白质 GST-EKL的诱导表达
表达质粒 p GEX-rEKL转化 E.coli BL21,37℃摇菌培养至菌 A600为 0.8左右,用终浓度为 0.8 mmol/L的 IPTG,25℃诱导表达 3 h。SDS-PAGE结果表明在Mr约 61 000处有特异表达条带,大小与推测的目的融合蛋白质 Mr大小相符合 (图 3)。发酵后的菌体经过超声破碎后分别取菌液上清、菌体、破碎上清和破碎沉淀做 SDS-PAGE,结果显示目的蛋白质为胞内表达,部分形成包涵体,部分以可溶形式存在 (图 4)。
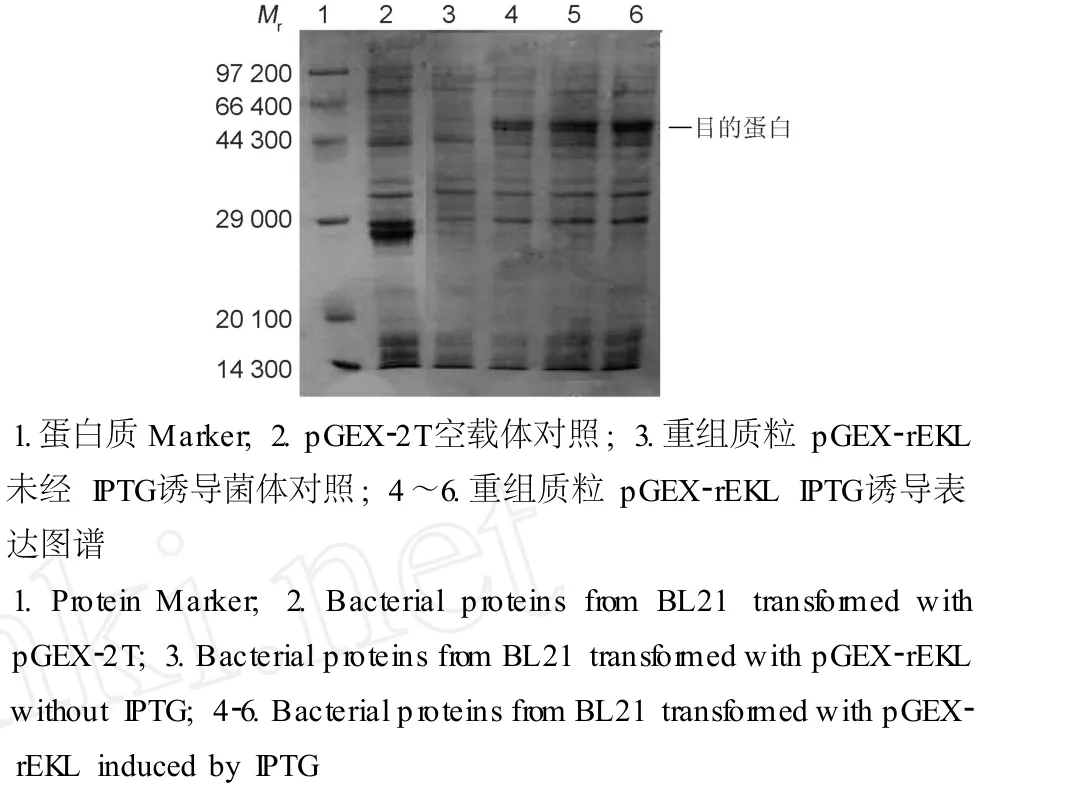
图3 融合蛋白质 GST-EKL的诱导表达分析Fig.3 SDS-PAGE analysisof induced GST-EKL expression
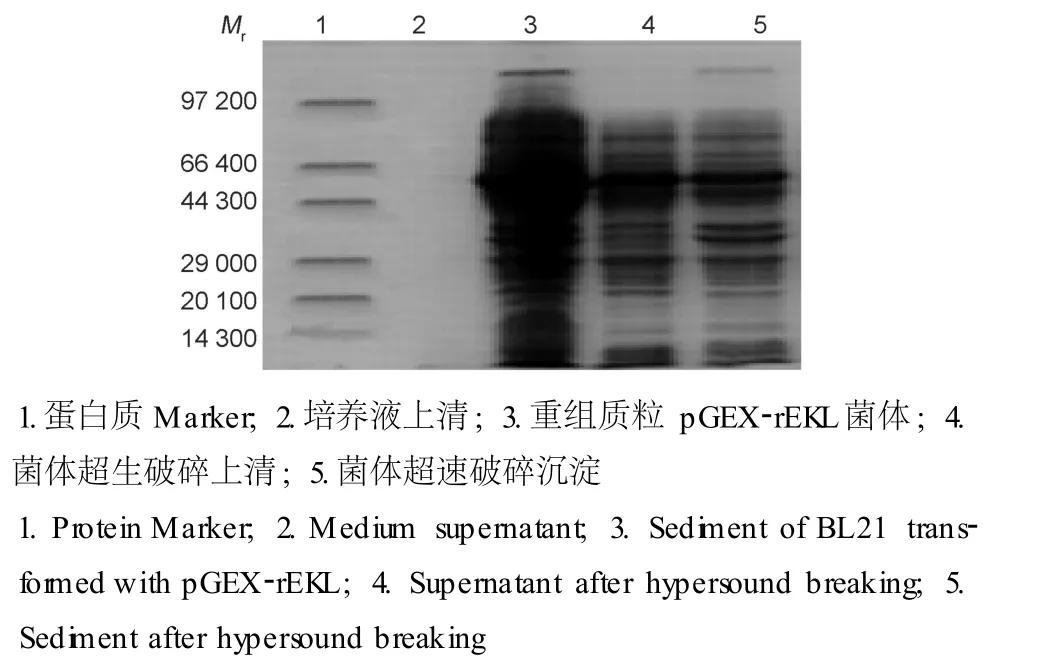
图4 超声破碎后融合蛋白质 GST-EKL的定位分析Fig.4 GST-EKL location analysis after hypersound breaking
2.3 融合蛋白 GST-EKL的纯化
将包涵体用尿素溶解后进行 SDS-PAGE检测结果显示包涵体中的蛋白质成功地被溶解纯化 (图5)。将含有融合蛋白质的清液通过 GST-Sepharose亲和色谱纯化后进行 SDS-PAGE检测结果显示融合蛋白质被成功纯化,洗脱峰呈现单一条带,Mr约为61 000(图6)。
2.4 融合蛋白质 GST-EKL的自切割及纯化
GST-Sepharose亲和色谱柱纯化后的 GST-EKL融合蛋白质用少量 EK切割引导,除去 GST标签,SDS-PAGE结果显示融合蛋白质成功被 EK切开,分为 GST标签和轻链 EK两部分 (图 7),而切割出的EK继续能切割体系中未完全切开的 GST-EKL融合蛋白,实现整个体系的自激活及切割,酶反应的混合物通过对氨基苯甲脒亲和色谱柱再次纯化,去除杂蛋白质,得到纯品。
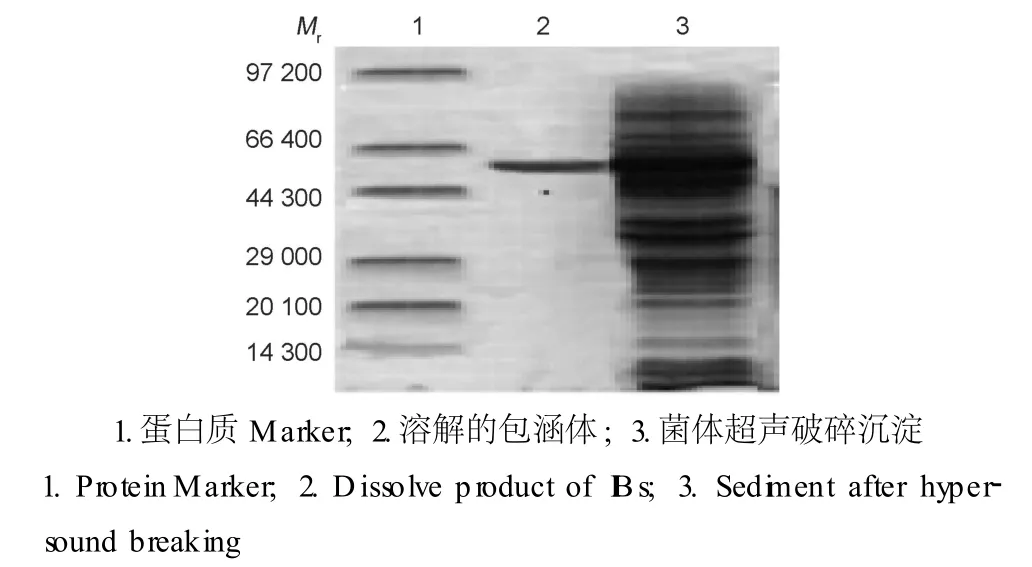
图5 包涵体尿素溶解的 SDS-PAGE分析Fig.5 SDS-PAGE analysisof IBsof GST-EKL
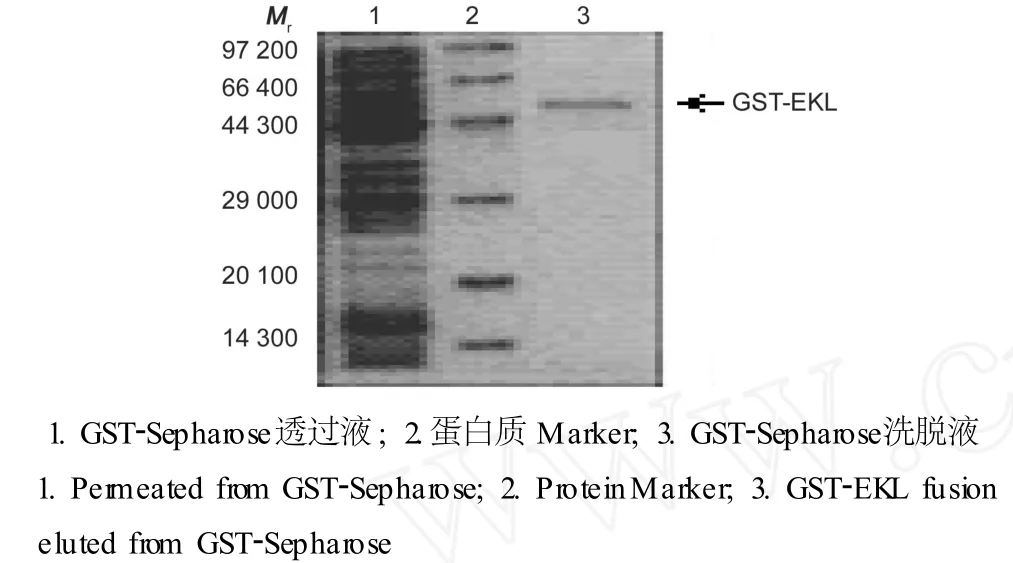
图6 融合蛋白质 GST-EKL的亲和色谱纯化Fig.6 Purification of the fusion protein GST-EKL
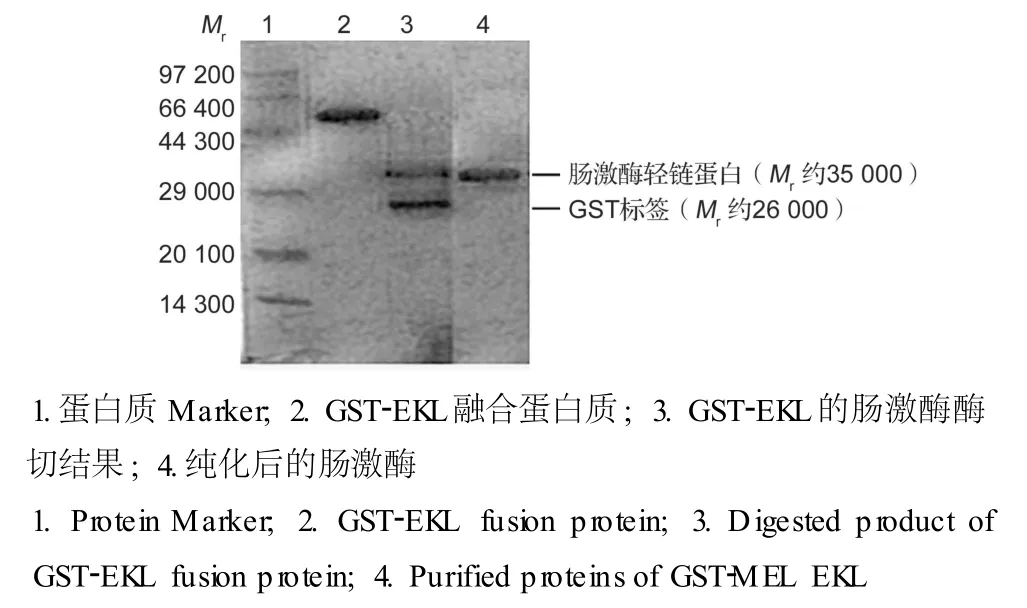
图7 融合蛋白质 GST-EKL的肠激酶引发自切割及对氨基苯甲脒亲和色谱Fig.7 SDS-PAGE analysis of GST-EKL by digestion with EK and purified p roteinsof EKL
2.5 重组 EK的活性鉴定
纯化后 E.coli表达的 EK浓度为 5.8 mg/mL,SDS-PAGE显示纯化后的 EK酶切效果好,在与融合蛋白质混合后,当酶与 GST-金黄色葡萄球菌蛋白 A比值为 1∶100时,目的融合蛋白质的被切割率在95%以上,结果见图 8。图 8出现了Mr26 000(谷胱甘肽硫转酶)和 32 000两条带,结果表明,我们制备出了具有生物活性的 EK。
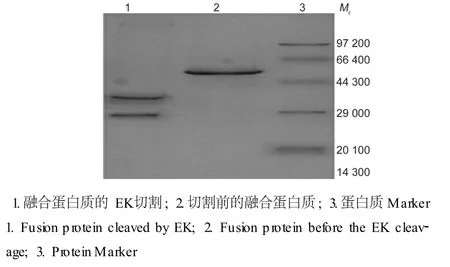
图8 表达的肠激酶对融合蛋白质酶切的 SDS-PAGE分析Fig.8 SDS-PAGE clevaging analysisof EK on fusion p rotein
3 讨 论
EK是胰蛋白酶原的生理激活剂,EK在体内激活胰蛋白酶原转化为胰蛋白酶,而胰蛋白酶是消化系统其它许多酶原的激活剂,因此 EK被认为是消化系统重要的起始酶之一[7]。一般认为 EK存在于小肠刷状缘细胞膜上,病理研究发现,十二指肠与胰腺的回流液中富集 EK会激活过多的胰蛋白酶原从而导致急性胰腺炎,而 EK缺陷的机体将会有腹泻、呕吐、浮肿等症状,造成发育不良,导致血蛋白不足症和贫血[8]。由于天然的 EK来源非常有限,本研究采用基因工程手段在原核表达载体中成功地表达和纯化了 EK轻链蛋白,可以解决前人纯化高成本、步骤繁琐的不足。
载体方面,表达载体用 p GEX-2T载体,其上具有 Plac启动子,能被乳糖类似物如 IPTG诱导,从而实现目的基因表达的严格调控和降低杂蛋白质本底,载体上带有 Amp的抗性基因,便于对重组阳性克隆菌的筛选,而且载体还自带 GST-tag,由于谷胱甘肽转移酶能够增加外源蛋白质的可溶性表达,所以我们将目的蛋白质构建在 GST标签蛋白下游来增加 EK的可溶性,同时带有 GST标签的融合蛋白质能通过 GST-Sepharose亲和色谱柱洗脱得到,我们已成功地应用该表达载体表达了多种生物活性蛋白[9-10]。
本研究成功的将 EK序列的 N末端引入 EK切割位点,表达了 EK轻链,在表达了 GST融合蛋白的同时,实现用少量 EK引发切割融合蛋白反应,致使EK产生与切割瀑布式放大,最终使整个体系完成自切割,可以得到具有天然 N末端氨基酸序列的 EK,由于 EK属丝氨酸蛋白酶,所以选择对氨基苯甲脒亲和色谱柱,除去杂质蛋白,得到高纯度的 EK蛋白,每 1 L培养基获得 EK约为 5.0 mg,简化了下游纯化工艺的难度,便于扩大化生产,促进了 EK在生物工程等领域的应用。
[1] 张向辉,谭树华,李泰明.肠激酶特点及其基因工程的研究进展[J].药物生物技术,2005,12(5):347-350.
[2] Mikhailova A G,Rumsh L D.Structural characteristics providing for high specificity of enteropeptidase[J].Bioorg Khim,1998,24(4):282-287.
[3] Light A,Janska H.The amino-terminal sequence of the catalytic subunit of bovine enterokinase[J].Protein Chem,1991,10(5):475-450.
[4] Yuan L D,Hua Z C.Expression,purification and characterization of a biologically active bovine enterokinase catalytic subunit in Escherichia coli[J].Protein Expr Purif,2002,25(2):300-304.
[5] Choi S I,Song H W,Moon JW,et al.Recombinant enterokinase light chain with affinity tag:expression from Saccharomyces cerevisiae and its utilities in fusion protein technology[J].Biotechnol Bioeng,2001,75(6):718-724.
[6] Tan Haidong,Wang Jinxia,Zhao Zongbao.Purification and refolding optimization of recombinant bovine enterokinase light chain overexpressed in Escherichia coli[J].Protein Expr Purif,2007,56(1):40-47.
[7] Lei F,Sun QM,Hua ZC.Expression of recombinant Chinese bovine enterokinase catalytic subunit in pastoris and its purification and characterization[J].Acta Biochimica Biophysica Sinica,2004,36(7):513-517.
[8] Light A,Janska H.Enterokinase(enteropeptidase):comparative aspects[J].Trends Biochem Sci,1989,14(3):110-112.
[9] 朱文赫,王 涵,孙妙囡,等.蜂毒明肽基因的原核表达及其纯化[J].中国生物制品学杂志,2009,22(10):971-974.
[10]朱文赫,田立玲,孙妙囡,等.蜂毒肽基因在大肠杆菌中的表达、纯化及初步鉴定[J].中国生化药物杂志,2010,31(1):1-5.
Clone and expression of bovine recombinant enterokinase catalytic subun it in Escherichia coli,self-activation cutting and assement assessm en t of b ioactivity
ZHU Wen-he1,GUO Zhi-gang2,LIU Hong-jian1,GUO Xiang-rui1,SUN De-jun1
(1.Department of Biomedicine,Institute of Frontier Medical Sciences,Jilin University,Changchun 130021,China;2.Orthopaedics,China-Japan Union Hospital of Jinlin,Jilin University,Changchun 130033,China)
Purpose The sequence of bovine EKL gene with Asp-A sp-Asp-A sp-Lys sequence in the N-terminus was obtained and analyzed,and the fusion protein of the EKL-GST was produced in E.coli.Methods The fragment of bovine enterokinase light chain cDNA was obtained by RT-PCR from bovine′s duodenal mucosa,then cloned into the pMD18-T cloning vector and sequenced.Compared with the sequence deposited in GenBank,the cloned gene sequence is correct.Then the interested gene fragment was inserted into the pGEX-2T expression plasmid.The recombinant vector pGEX-rEKL was transformed into E.coli BL21 and induced by IPTG.Results SDS-PAGE analysis indicated that the target product was about 61 kDa.The research successfully clones and expresses EKL-GST fusion protein in E.coli.Selt-activation by microamount cutting the fusion protein,and purified the EKL by minobenzene-Sepharose.Conclusion This investigation would be able to lay a foundation for enterokinase activity research and further application of expression products on a large scale.
enterokinase;Escherichia coli;expression system;GST self-activation
Q78
A
1005-1678(2011)01-0006-05
2010-01-28
吉林省科技发展计划项目(20060564)
朱文赫,男,博士研究生;孙德军,通信作者,Tel:0431-85619233,E-mail:sundjjl@yahoo.com.cn。
