化学新药研发企业对临床试验用药品委托制备的研发质量管理实践
2024-02-27陈辉刘俊耀夏广新
陈辉 刘俊耀 夏广新
摘 要 临床试验用药品是化学新药研究与试验发展(以下简称研发)过程中里程碑式的产品,其制备的质量直接影响到临床受试者的用药安全性和临床试验结果的有效性。为确保临床试验用药品在委托制备过程中安全、有效且质量可控,本文阐明了化学新药药学研究在不同阶段下的研究重点,深入分析了工作中对临床试验用药品委托制备的质量管理所遇到的问题,探索出以“技术转出资料管理”“委托合同”“质量协议”三位一体,协同对临床试验用药品委托制备研发质量管理的模式,以期为新药研发企业的研发质量管理提供借鉴与思考。
关键词 化学新药 临床试验用药品 委托合同 质量协议 技术转出资料管理
中图分类号:R951 文献标志码:C 文章编号:1006-1533(2024)01-0065-04
引用本文 陈辉, 刘俊耀, 夏广新. 化学新药研发企业对临床试验用药品委托制备的研发质量管理实践[J]. 上海医药, 2024, 45(1): 65-68; 79.
R&D quality management practice for commissioned preparation of investigational medicinal products in chemical new drug R&D enterprise
CHEN Hui, LIU Junyao, XIA Guangxin
(Central Research Institute, Shanghai Pharmaceuticals Holding Co., Ltd., Shanghai 201203, China)
ABSTRACT Investigational medicinal products are milestone products in the research and experimental development(R&D) of new chemical drug, and the quality of their preparation can directly affect the safety of clinical subjects and the validity of clinical trial results. In order to ensure the safety, effectiveness and controllable quality of investigational medicinal products in the process of commissioned preparation, this article clarifies the research emphases of pharmaceutical research on new chemical drugs in different stages and analyses the problems encountered in the quality management of commissioned preparation of investigational medicinal products, explores a trinity mode of “technology transfer data management”, “commissioning contract”and “quality agreement” to cooperate with the R&D quality management of commissioned preparation of investigational medicinal products and to provide reference for the R&D quality management of chemical new drug R&D enterprises.
KEY WORDS chemical new drug; investigational medicinal products; commission contract; quality agreement; technology transfer data management
臨床试验用药品的制备具有特殊性[1]。在早期临床试验阶段,原料药/制剂的工艺和质量控制存在很多不确定性,理化及生物学特性等关键质量属性研究不充分,于此建立的质量控制策略,质量标准及检验操作规程也较宽泛。同时,临床试验设计的多样性对生产、包装与贴签提出更多的要求[2],这必然使得对临床试验用药品的研究与试验发展(以下简称研发)的质量管理不同于商业化药品《药品生产质量管理规范(good manufacturing practice, GMP)(2010年修订)》的管理模式,从而对新药研发企业的研发质量管理提出了很大的挑战。为此,须构建覆盖化学新药研发全过程的研发质量管理体系,遵循化学新药研究所具有的渐进性、阶段性、不确定性的特点,扎实地开展研发质量管理工作,管控研发质量风险,解决委托过程中遇到的问题,确保临床试验用药品的安全、有效、质量可控。1 本文所述概念的界定
1)化学新药是指Ⅰ类化学新药,根据国家药品监督管理局发布的《化学药品注册分类及申报资料要求》中所述,是在境内外均未上市的创新药,指含有新的结构明确的、具有药理作用的化合物,并且具有临床价值的药品[3]。
2)临床试验用药品是指用于临床试验中的试验药物、对照药品、安慰剂[2]。
3)技术转出资料:化学新药委外制备时,为了确保对原料药/制剂的认知能准确转移,及所满足的质量要求,委托方向受托方提供原料药/制剂的工艺和质量控制要求、理化及生物学特性等知识合集。
4)委托方是指上海医药集团股份有限公司中央研究院(简称:上药研究院),负责临床试验用药品的制备工作。
5)受托方是指受托制备临床试验用原料药/制剂的企业。
2 国内关于临床试验用药品的法律法规
国家药品监督管理局为保证临床试验用药品的安全、有效和质量可控,施行的相关法律法规中,对临床试验用药品的规定有:
《中华人民共和国药品管理法》中第十九条规定:开展药物临床试验,应当按照国务院药品监督管理部门的规定如实报送研制方法、质量指标、药理及毒理试验结果等有关数据、资料和样品,经国务院药品监督管理部门批准[4]。《药品注册管理办法》中规定第二十五条规定:药物临床试验用药品的管理应当符合药物临床试验质量管理规范的有关要求[5]。《临床试验用药品》附录中第二条规定:临床试验用药品的制备应当遵循GMP的相关基本原则以及数据可靠性要求[1]。
至此,国家药品监督管理局明确了我国对临床试验用药品制备的质量管理要求,为新药研发企业及受托生产企业对于临床样品制备的合规性管理,提供了制度保障;同时也考虑临床试验阶段的特殊性,采取适当的质量风险控制策略,降低风险,确保临床试验用藥质量,最大程度保障受试者的安全。
3 化学新药药学研究在不同阶段下的研究重点
化学新药研发是一个多学科、强合规、长周期的探索过程,具体包含药物发现阶段、临床试验申报(investigational new drug, IND)阶段、临床试验研究阶段和新药上市申报(new drug application, NDA)阶段。而化学新药的药学研究具有渐进性、阶段性和不确定性特点,其研究的广度和深度伴随临床试验进展不断推进[6]。
3.1 临床试验申报阶段的研究重点
在临床试验申报阶段,开展药物的化学、制造和控制(chemical manufacturing and control, CMC)研究,就考虑成本和时效来说,必然追求以快速成功递交IND申报资料,并获得临床批准通知书为首要目标。
对于原料药研发工作而言,这一阶段须快速开发出一条适合放大的合成路线,确认当下所认知的关键工艺参数和关键工艺步骤,以制备用于安全性研究以及临床申报的原料药;与此同时,进行分析方法开发,对关键物料、中间体、原料药进行检测,积累数据,拟定关键物料、中间体、原料药研究质量标准,初步确定原料药的有关物质、溶剂残留、含量等分析方法,并开展至少包括专属性、灵敏度等关键项的分析方法验证和通用方法的确认。这一切都是在遵循科学规律且满足注册法规要求下抢占先机,对原料药理化特性和杂质行为等关键质量属性因研究不深而了解有限。
对于制剂而言,这一阶段基于对药物的理化特性、安全性和有效性的有限了解,通常开发处方工艺不复杂,研制的剂型比较简单(多为口服片剂或者胶囊),以方便剂量探索,此阶段的剂型和处方工艺还存在很大的不确定性。与此同时,开发制剂的分析方法,并对中间产品、制剂成品进行检测及数据汇总,拟定中间产品、制剂成品的研究质量标准及检验方法,确定用于制剂成品的分析方法,开展至少包括专属性、灵敏度等关键项的分析方法验证,基于有限的稳定性、相容性等研究信息初步确定制剂的包装、贮藏条件。
3.2 临床试验阶段研究重点
进入临床试验研究阶段,根据国内法律法规的要求,须在GMP下制备满足临床试验需求用的药品。这个阶段是对原料药/制剂认识不断加深,逐步对合成工艺/处方工艺、分析方法、质量标准等进行调整优化的过程。
这一阶段,对于原料药研究工作,基于对关键工艺参数和关键质量属性的深入理解,不断优化制备工艺,基本确定杂质谱,确保制备出的不同批次间的原料药质量一致。同时,确定关键物料、中控、中间体、原料药研究质量标准及其检验操作规程,随着临床研究的深入,至少完成关键物料和原料药的方法学验证或通用方法的确认,还需完成清洁验证和残留物检测方法的验证,确保方法的检测能力可行、检测数据可靠。
这一阶段,对于制剂而言,确定临床样品单位剂量的处方组成及工艺规程,基本确定杂质谱,确保制备出的不同批次间的制剂质量一致。同时,确定辅料、直接接触药品的包装材料和容器的质量标准及检验操作规程;随着临床研究的深入,确定制剂中间产品及成品的质量标准及其检验操作规程,开展方法学验证和通用方法的确认;此外,也应完成清洁验证和残留物检测方法的验证,确保方法的可行性、有效性、可靠性。
4 上药研究院对临床试验用药品委托制备的研发质量管理实践
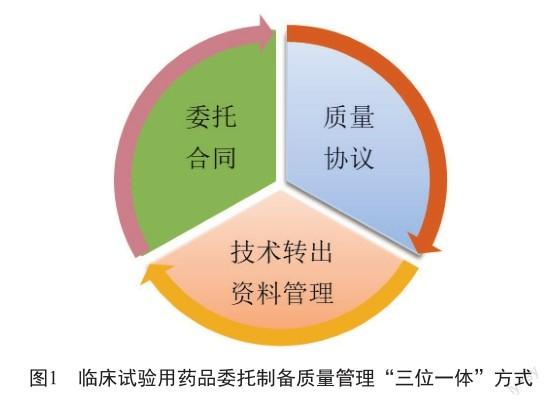
上药研究院制备临床试验用药品,采用委外制备。上药研究院研发质量管理团队在实践中构建覆盖了从药物发现、IND阶段、临床试验、NDA阶段的研发全过程及多学科的研发质量管理体系,探索出以“技术转出资料管理”“委托合同”“质量协议”为三位一体,协同对临床试验用药品委托制备质量管理的模式(图1)。其中,“技术转出资料管理”规范了技术转出资料的完整性与合规性要求,是委托制备成功的基石;“委托合同”则是约定委托方和受托方各自承担责任的法律保障,在合同中约定了质量管理要求;而“质量协议”则是管控研发质量风险,解决委托过程中遇到的质量问题,保证委托制备全过程持续符合法定要求的抓手,从而保障临床试验用药品安全、有效、质量可控。
4.1 技术转出资料管理
在委托制备过程中,对技术转出资料的管理是委托过程的关键环节,关系到产品持续稳定制备,质量持续可控。委托制备临床试验用药品时,须编写技术转出资料,参考CTD(common technical document, 通用技术文件)格式和要求,依据在临床试验申报阶段已掌握的原料药/制剂的认知,诸如原料药及制剂的工艺知识、物料及其供应商信息、质量研究知识的内容进行总结,编写技术转出资料。
其中对于工艺知识及其质量研究知识方面,在管理中经常遇到诸如在工艺描述中,对杂质的产生、转化、清除过程的研究不够充分,对杂质控制的策略描述不够详细;还有如未对包材的选择、包装要求作出说明,而导致后续的原料药稳定性研究时出现水分检测超标现象。本团队针对这些问题,在工艺知识内容方面,强调写明包装工序中内包材的质量标准、供应商信息(及标签)和包装要求;在质量研究知识方面,强调写明杂质的信息如检验报告、储存条件、杂质目录。如此,便确保了技术资料的完整性。
其中,对于物料及其供应商的管理,本团队曾遇到供应商拒绝提供物料的质量标准及检测方法和检验报告,且拒绝接受现场审计,而研发人员坚持该供应商提供的物料最符合工艺要求,最终导致在开展工艺验证时,无法对该供应商进行管理。此外,在不同阶段下对物料的检测要求也不一样,如在临床试验早期阶段,可凭供应商提供的检测报告进行鉴别或核对后即可使用,无须对每个物料检验;而在开展工艺验证时,则要求所有物料须全部检验合格后放行使用。为了保证物料管理的延续性、合规性要求,本团队提出将所用物料,依据其放行风险分成三类:
第一类物料是上药研究院制定质量标准的物料,如关键物料、工艺中特殊需求的试剂、溶劑、催化剂等,这类物料由上药研究院提供质量标准及检测方法;第二类物料是采用供应商质量标准的物料,如原料药制备用的试剂、溶剂、催化剂、包材等,制剂制备用的辅料、专用溶剂、包装材料等,也包括具有剧毒等特殊属性或现场抽样检测困难的物料、辅料、直接接触药品的包装材料或容器等,这类物料需委托方从供应商处索取物料的质量标准、检测方法及检验报告,提供给受托方;第三类物料是采用受托方提供的纳入其GMP体系的质量标准类物料,如通用试剂、溶剂等物料,这类物料的质量标准及检测方法及检验报告,授权给受托方负责。这样形成对物料的差异化管理,在临床试验早期阶段,只需按三类物料的质量标准要求,对应进行检测、复核、检验,合格后即可放行使用;而在开展工艺验证时,对第一类和第三类物料按其质量标准检验合格后即可放行使用,对于第二类物料,通过对其检测方法及检测项目的累计数据进行评估,确定其质量标准及检验操作规程,进而检验合格后放行使用。使用的所有物料中具有剧毒等特殊属性或现场抽样检测困难的物料、辅料、直接接触药品的包装材料或容器等,则可通过对供应商的审计,确定其为合格供应商后,即可凭供应商的检验报告放行。如此一来,在写技术资料时,强调写明物料的类别及其供应商信息,对于不予配合的供应商,也可及时更换,避免出现后续管理困难。
对于物料供应商的管理,本团队则采取部分授权给委托方负责。如对供应商的评估与选择,授权受托方对所用物料的供货厂商,依据其所提供的物料检验结果、质量投诉、不合格处理记录等进行质量回顾分析,并提供回顾分析报告给委托方审核。委托方认可回顾分析报告后,据此对物料供应商进行质量评估,对质量评估不符合要求的供应商行使否决权。而对物料供应商的质量审计,由委托方负责,受托方配合共同参与,将通过审计的厂商,列入合格供应商目录。
针对上述管理中遇到的问题,本团队还强化在转出前对技术资料内容的审核,关注工艺描述中对关键工艺的步骤、参数是否确认;对于返工操作,是否明确返工工艺、次数;对于后处理的浓缩或者烘料操作,是否写明终点判断及检测方法;对于不同类别物料的质量标准及其检测方法、检验报告是否完整等。在审核相关证明材料时,关注物料供应商的信息是否完整,重点关注供应商提供的物料使用授权证书、物料的质量标准及其分析方法、检验报告单是否提供齐全。由此,本团队通过对技术转出资料的审核,确保了技术转出资料内容的完整性、合规性要求,从而保障对原料药/制剂的认知能准确转移。
4.2 委托制备合同的质量管理要求
委托制备须签订委托制备合同,这是落实双方各自责任的法律保障。为此,本团队联合法务部门一同编制了为临床试验用药品委托制备专用的委托合同模板,在合同中约定了质量管理要求。
委托制备原料药/制剂任务,在合同中约定受托方应满足受托项目所需的GMP要求,提供与受托项目相匹配的足够数量,且合格的人员、厂房、设施和设备;制备的原料药/制剂应符合委托方与受托方约定的质量标准;对于所得量,原料药规定制备批次、最终所得量;和制剂成品的批次、最终入库量,确保满足临床试验用。同时规定受托方确保过程,所有记录遵循真实、准确、完整、可追溯原则。
对于在临床试验申报阶段,受制于研发条件而尚未开展微生物检测方法开发、对直接接触产品的生产设备和用具的清洁操作和开发残留物检测方法的研究任务,须在临床试验阶段完成。对此,在合同中约定委托受托方在GMP下,进行研究和技术开发,且约定受托方在完成研究任务后,负责编写相应工艺规程、质量标准及检验操作规程,并书面呈送给委托方审核、确认后,受托方批准执行。
委托制备过程,质量审计是有效管控质量风险的重要管理方式。对于质量审计的要求,在合同中约定委托方有权对受托方制备现场进行质量审计,受托方对审计发现的缺陷应及时整改,在规定时限内将整改报告上报给委托方,并得到委托方认可;委托方在接受药品监管部门监督检查时,如需提供委托研究相关资料的,受托方应当配合提供;需要开展现场检查时,受托方应当予以配合。
团队通过对合同中质量管理要求的约定,形成委托方和受托方共同遵守和维护的基本准则,从而实现对委托制备全过程的质量合规把控。
4.3 质量协议的质量管理要求
在《临床试验用药品(试行)》中第六条中,明确规定,对于临床试验用药品委托制备,双方需签订质量协议[1]。为此,本团队专门编制了为临床试验用药品委托制备专用的质量协议模板,作为委托制备合同的附件。
按国家法规要求,临床申请人对临床试验用药品的质量承担全部责任;又面临着药学研究所具有的渐进性、阶段性和不确定性特点,为了管控研发质量风险,本团队积极开展委托制备的质量审计工作,对受托方进行委托前、委托中和委托后的质量审计,实现对委托制备全过程的质量合规把控。
委托制备前,对于首次委托制备临床试验用药品的受托方,本团队积极开展对受托方委托制备前的质量审计工作,对受托方的生产条件、技术水平和质量管理情况进行评估,确认受托方具有受托生产的条件和能力,能持续符合GMP以及委托生产产品的生产质量管理要求。考察通过后,委托方与受托方签订合同及质量协议(包括保密协议)后,委托方向受托方提供委托生产药品的且通过本团队审核过的技术转出资料,约定受托方应依据委托方提供的技术转出资料,编制工艺规程,质量标准及检验标准操作规程,稳定性试验方案等文件,并书面呈送给委托方审核,重点关注受托方的数据信息跟委托方转出的数据信息相一致,给出审核意见,并书面反馈给受托方。由此,加强了委托方、受托方间的沟通,使得受托方深入理解项目的需求,团队也起到了在过程中管理的作用。
委托制备过程中,由于药学研究的渐进性、阶段性、不确定性特点,偏差与变更是必然事件,但这不同于商业化药品GMP管理模式。为此,团队对于偏差、变更的管理,会依据风险管控原则,对于重大偏差/变更及时跟踪审核把关,必要时进行现场审计管理的方法:规定受托方与委托方应基于研究数据进行风险评估,分析根本原因,将涉及重大偏差/重大变更的文件书面呈送给委托方审核;委托方给出审核意见,并书面反馈给受托方;受托方按审核意见执行;受托方在重大偏差/变更关闭后,将其报告发送给委托方评估实施效果。而对于一般偏差/变更,则授权给受托方按规程管理,且受托方在产品出厂放行后,将其报告发送给委托方审核。委托制备结束后,团队对受托方制备现场开展质量审计,审计通过后,以便委托方批准临床药品放行,确保临床药品的安全、有效、质量可控负责。与此同时,在质量协议中也明确了药品档案管理,规定受托方在制备完成后,需提供项目相关的资料给委托方,从而有利于委托方对整个项目进行回顾性管理。
5 总结与展望
临床试验用药品是化学新药研发过程中里程碑式的产品,其制备的质量可控性直接影响临床受试者的用药安全性和临床试验结果有效性的判断,进而关系到对化学新药的评价和上市价值。
从新药研发企业内部而言,进一步完善新药研发质量管理体系,夯实“三位一体”的管理模式,遵循新药研究所具有的渐进性、阶段性、不确定性的特点,强化研发风险管控,并不断推广到对生物药、中药的管理中,扎实地开展研发质量管理工作,全面提升企业对临床试验阶段委托制备的合规性管理水平。
从药品监管部门的角度而言,针对临床试验用药品的管理,应进一步理清关键问题,比如在临床药品制备中,设置了“临床药品放行责任人”,这与质量负责人、质量授权人的法律地位的定位又应如何划分等问题。针对种种情况与问题,药品监管部门应进一步明确,切实加强药品全生命周期管理,充分保证临床试验用药品的安全、有效和质量可控,最大程度保障受试者的安全。
參考文献
[1] 国家药品监督管理局. 国家药监局关于发布《药品生产质量管理规范(2010年修订)》临床试验用药品附录的公告(2022年第43号)[EB/OL]. (2022-05-27)[2023-02-06]. https:// www.nmpa.gov.cn/xxgk/fgwj/xzhgfxwj/20220527182006196. html.
[2] 许丹, 王元, 张毅敏, 等. 临床试验用药品生产质量管理的初步探讨[J]. 中国新药杂志, 2021, 30(18): 1649-1654.
[3] 国家药品监督管理局. 国家药监局关于发布化学药品注册分类及申报资料要求的通告(2020年第44号)[EB/OL].(2020-06-29)[2023-02-06]. https://www.nmpa.gov.cn/xxgk/ ggtg/ypggtg/ypqtggtg/20200630180301525.html.
[4] 中华人民共和国药品管理法[L/OL]. (2019-08-27)[2023-02-06]. https://www.nmpa.gov.cn/xxgk/fgwj/flxzhfg/ 20190827083801685.html.
[5] 国家市场监督管理总局. 药品注册管理办法[EB/ OL]. (2020-01-22)[2023-02-06]. https://www. samr.gov.cn/zw/zfxxgk/fdzdgknr/fgs/art/2023/art_ 3275cb2a929d4c34ac8c0421b2a9c257.html.
[6] 国家药品监督管理局药品审评中心. 创新药(化学药)临床试验期间药学变更技术指导原则(试行)[EB/OL].(2021-03-12)[2023-02-06]. https://www.cde.org.cn/zdyz/dom esticinfopage?zdyzIdCODE=e1a358fd48693f910793b109794 d55ab.
