手性钌螯合催化剂的合成及在α—羟基酯的动态动力学氢化反应中的应用
2020-10-16汪贲文汤卫军
汪贲文,杜 甜,汤卫军
(1.陕西师范大学化学化工学院,西安710119;2.兰州交通大学实验管理中心,兰州730070)
酯类化合物广泛存在于日常生活中.酯还原为醇是一个重要的还原反应,常用的还原剂有氢化铝锂和硼氢化钠等.近年来,随着催化化学的发展,酯的氢化还原反应研究取得了显著的进展[1~4],相应的高效催化体系被开发出来,羰基氯化氢化[双(2-二苯基膦乙基)氨基]钌催化剂(Ru-MACHO)[5]已成功应用于氢化还原手性乳酸甲酯为手性1,2-丙二醇化合物的工业化生产.最近,本课题组[6]利用该催化剂实现了α-酮酸酯选择性氢化还原生成α-羟基酯或1,2-二醇化合物的反应,特别是α-羟基酯化合物,为进一步的官能团衍生提供了可能.
动态动力学拆分是合成手性分子的重要方法[7~9],其反应实质是在手性催化剂的作用下,一对消旋对映异构体最终成为单一对映异构体化合物.1989年,Noyori等[10]在均相催化氢化条件下对α-乙酰胺-β-酮酸酯进行了动态动力学拆分还原,最高可以达到98%的对映选择性,因此该方法被广泛应用于氢化及氢转移反应中,并逐渐成为获取手性化合物的有效方法之一[11,12].目前,动力学拆分还原反应的底物适用范围主要集中在α-取代酮(醛)[13~18]、取代-β-酮酸酯或酰胺[19~23]、α-取代-β-酮磷酸酯或砜[24,25]和亚胺衍生物[26~31]. 但针对α-取代酯的动态动力学拆分的研究报道较少.Chi等[32,33]采用一类卡宾催化剂通过酯交换的模式实现了α-取代酯的动态动力学拆分,其对映体过量值(e.e.)最高可达98%.2018年,Tokunaga等[34]通过相转移催化,将α-保护氨基的酯类化合物动态动力学拆分水解成酸.Zhang[35]及Bergens等[36]通过氢化还原酯基,在氢化还原内酯及α-芳基醚酯上实现了动态动力学拆分,均取得较好的结果.近年来,基于螯合型金属催化剂氢化还原酯类化合物的研究取得了很大进展,合成了一类螯合型手性催化剂并探讨相应的手性识别能力,这将有助于促进该研究领域的发展.
本文设计合成了一类手性螯合配体,与二氯三(三苯基磷)合钌[Ru(PPh3)3Cl2]反应得到相应的钌螯合配合物,对其进行了结构表征.将该催化剂应用于非保护α-羟基酯类化合物的动态动力学氢化反应,通过优化反应条件实现了酯类的氢化还原,并得到中等强度的对映选择性,为设计高效的催化体系奠定了研究基础.
1 实验部分
1.1 试剂与仪器
2-吡啶甲醛、5-Br-2-吡啶甲醛和5-甲基-2-吡啶甲醛均购于毕得医药有限公司;(R)-2-二苯基膦-1-苯基乙胺、(R)-2-二苯基膦-1-苄基乙胺、Ru(PPh3)3Cl2、扁桃酸甲酯、甲醇钾(KOMe)、叔丁醇钾及正丁基锂购于泰坦生物化学试剂有限公司;甲醇、甲苯、乙醚、四氢呋喃、丙酮、1,4-二氧六环、1,8-二氮杂二环十一碳-7-烯(DBU)及碳酸钾等试剂购于国药集团化学试剂有限公司;所有试剂均为分析纯.所用溶剂均经无水无氧处理后使用.参照文献方法分别合成3-Br-2-吡啶甲醛及异喹啉-1-甲醛[37]、5-甲氧基-2-吡啶甲醛及5-异丙氧基-2-吡啶甲醛[38];参照文献[39]方法合成5-(1-环戊基亚胺基)-2-吡啶甲醛和5-(1-环己基亚胺基)-2-吡啶甲醛.
Bruker AscendTM400 MHz型核磁共振波谱(NMR)仪,瑞士Bruker公司;ApexⅣFTMS型高分辨质谱(HRMS)仪,电喷雾(ESI)源,瑞士Bruker公司;LC-2010AHT型高效液相色谱(HPLC)仪,日本岛津公司.
1.2 实验过程
1.2.1 配体的合成 参照文献[40]方法,将1.2 g 2-吡啶甲醛及其衍生物与3.05 g(R)-2-二苯基膦-1-苯基乙胺进行缩合及还原反应,合成相应的配体(见Scheme 1),经柱色谱分离,得到3.66 g淡黄色油状产物,收率93%.1H NMR(400 MHz,CDCl3),δ:8.41(d,J=4.4 Hz,1H),7.48(t,J=4.7 Hz,1H),7.31~7.24(m,5H),7.21~7.12(m,12H),7.03~6.97(m,2H),3.65~3.52(m,3H),2.48~2.37(m,2H);13C NMR(100 MHz,CDCl3),δ:158.7,148.2,143.0(d,J=5.5 Hz),137.4(q,J=12.4 Hz),135.1,131.8(q,J=19.1 Hz),127.5,127.4,127.4,127.4,127.3,127.3,126.3,126.1,121.2,120.7,59.0(d,J=15.5 Hz),51.9,37.7(d,J=14.2 Hz);31P NMR(162 MHz,CDCl3),δ:-22.9(d,J=6.4 Hz,1P);HRMS(C26H25N2P计算值),m/z:397.1822(397.1828)[M+H]+.

Scheme 1 Synthesis of ligands
1.2.2 催化剂的合成 参照文献[41]方法,将上述得到的配体与Ru(PPh3)3Cl2反应获得催化剂Cat.1~Cat.12(Scheme 2),其理化性质、高分辨率质谱及核磁共振波谱数据分别列于表1和表2.

Scheme 2 Synthesis of catalysts
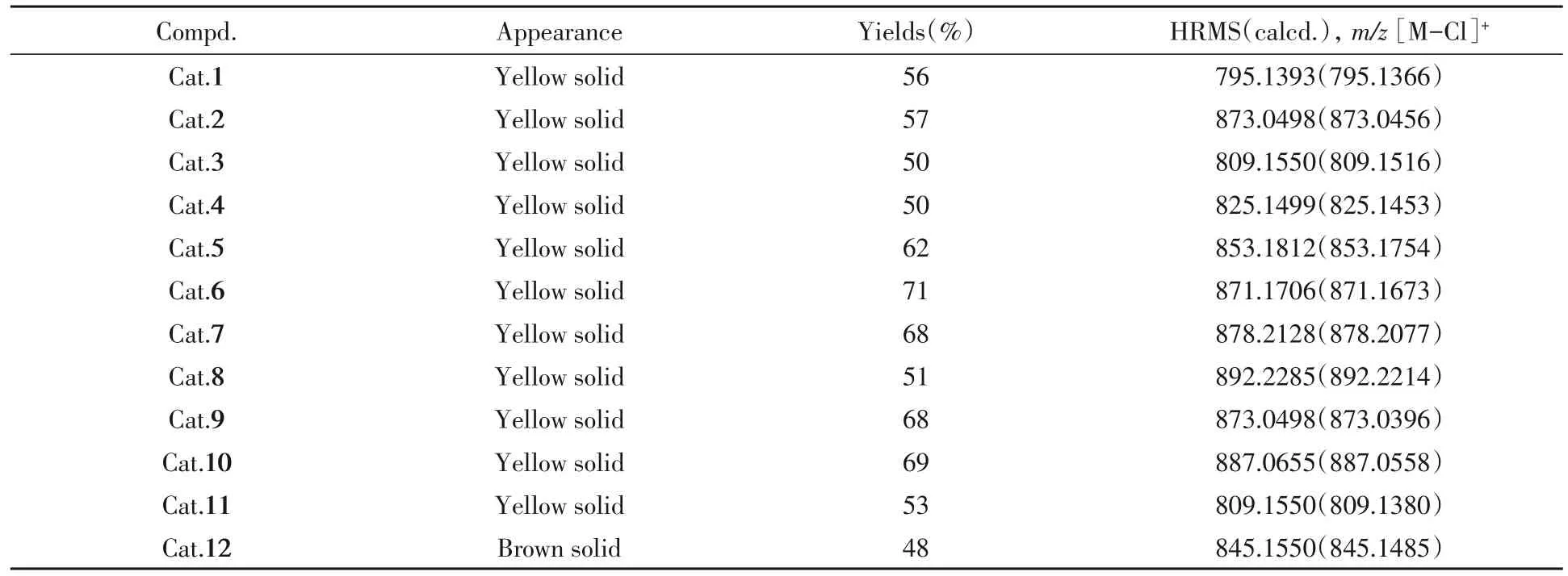
Table 1 Appearance,yields and HRMS data of compounds Cat.1—Cat.12

Table 2 1H NMR,13C NMR and31P NMR data of compounds Cat.1—Cat.12

Continued
1.2.3 晶体结构的表征 为了确定催化剂的结构,选取Cat.5进行单晶的培养,测定其晶体结构.由Scheme 3可见,钌金属上2个氯原子处于反式位置,与非手性的钌螯合化合物具有类似的结构[41],有关晶体数据可访问www.ccdc.cam.ac.uk,编号:2006158.
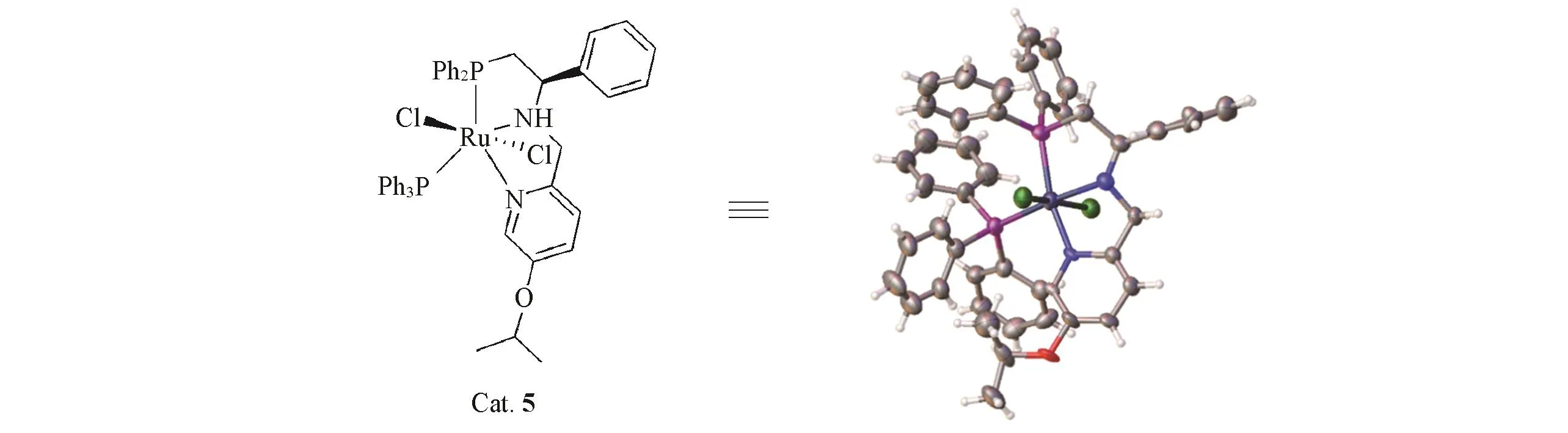
Scheme 3 Crystal structure of Cat.5
1.2.4 以α-羟基酯为底物的动态动力学氢化反应 参照文献[41]方法,以α-羟基酯为反应底物,在手套箱中将 41.5 mg(0.15 mmol)扁桃酸甲酯、1.3 mg(0.0015 mmol)催化剂 Cat.1、9 mg(0.125 mmol)KOMe和700 μL四氢呋喃依次加入到反应玻璃管中,将玻璃管放入高压釜中,充入5 MPa氢气,于40°C反应12 h,α-羟基酯全部转化为二醇产物,产物的e.e.值为44%.
2 结果与讨论
2.1 反应条件的筛选
在初步的反应中,取得了一定的对映选择性,这主要归因于催化剂与底物存在一定的手性匹配作用,表现出不同的反应速率.同时,由于底物中酯基的吸电子诱导作用,在强碱作用下α-氢容易发生消旋化,从而实现动态动力学拆分.对照实验也表明,手性扁桃酸甲酯和甲醇钾在四氢呋喃溶液中搅拌1 h,得到消旋的扁桃酸甲酯.为了获得最佳反应条件,分别对酯基类型、碱的用量、反应温度、反应时间及反应溶剂进行了筛选,结果列于表3.
由表3可见,不同的酯基取代基对反应有一定的影响.将甲基换成乙基,e.e.值略有下降;换成异丙基,反应几乎不发生;换成苄基,得到中等的转化率和53%的e.e.值(表3中Entries 1~4).增加或减少碱的用量,均降低了反应的对映选择性(表3中Entries 5和6).降低反应温度至室温,可以保持相似的反应活性及对映选择性(表3中Entry 7);继续降低温度不利于原料转化(表3中Entry 8).考虑到催化剂在室温具有较高催化活性,在保证相同转化率的情况下,缩短反应时间时e.e.值可提高至51%(表3中Entry 9);进一步缩短反应时间尽管对e.e.值提高稍有影响,但降低了原料的转化率.后续研究发现,碱对反应具有重要影响,使用其它无机强碱和弱碱,如甲醇钠、乙醇钠、氢氧化钾、氢氧化钠及碳酸铯等,均不发生反应,只有叔丁醇钾获得99%的转化及44%的e.e.值(表3中Entry 13).这说明碱不仅协助氢气的裂解,对底物的消旋化速度也有重要影响,碱的强度与浓度均影响对映选择性.此外,不同的溶剂也会影响反应的速度及e.e.值(表3中Entries 14~17),使用二氯甲烷或1,4-二氧六环,虽然可使底物全部转化,但对映选择性却比四氢呋喃低;使用甲苯可获得57%的e.e.值,转化率却偏低.采用极性质子性的甲醇溶剂,催化活性及对映选择性均显著降低,并得到构型翻转的产物(表3中Entry 17).
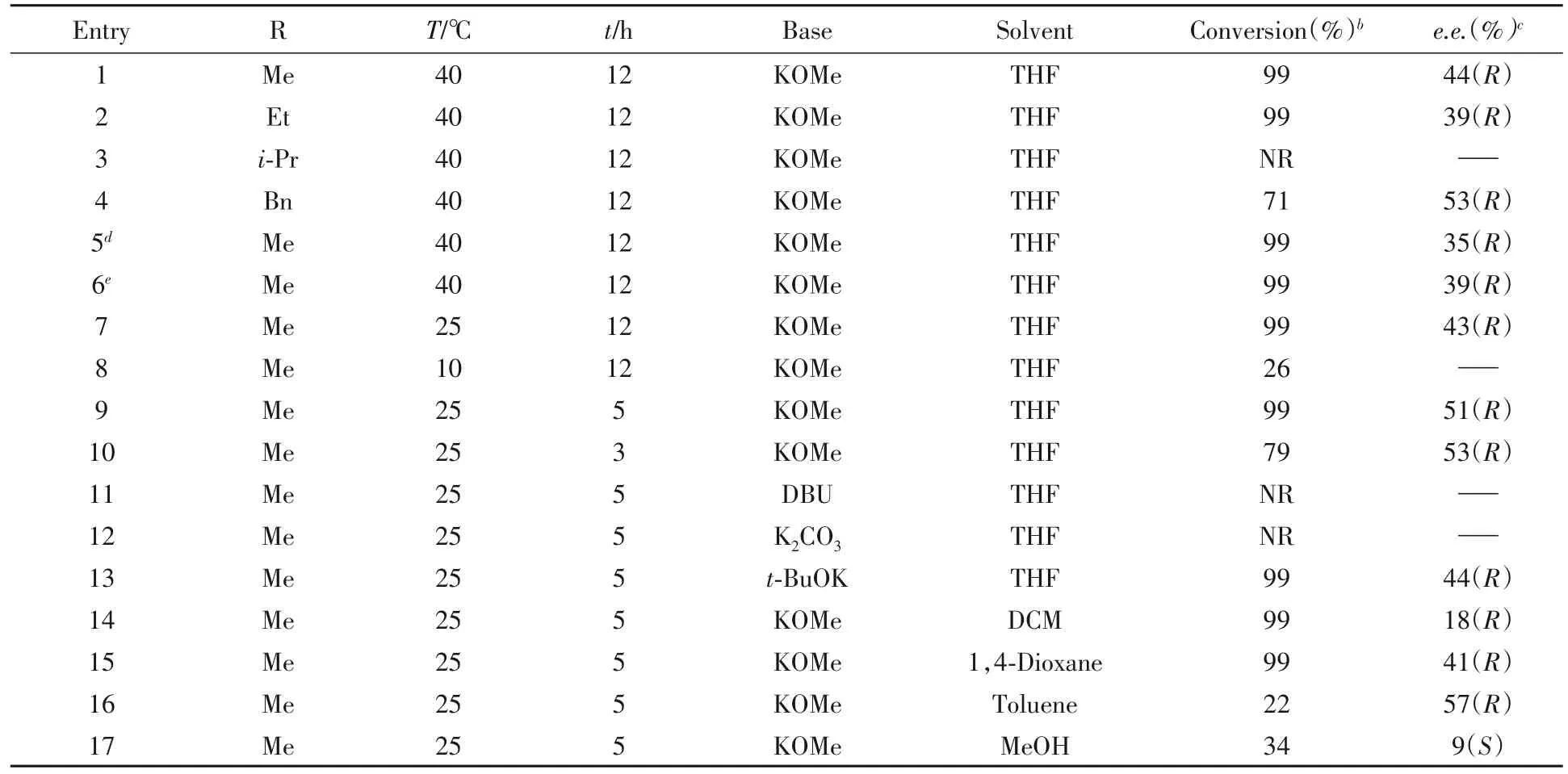
Table 3 Optimization of reaction conditionsa
2.2 催化剂的筛选
根据初步筛选结果,对另外11种含不同取代基的催化剂进行了筛选,结果列于表4.
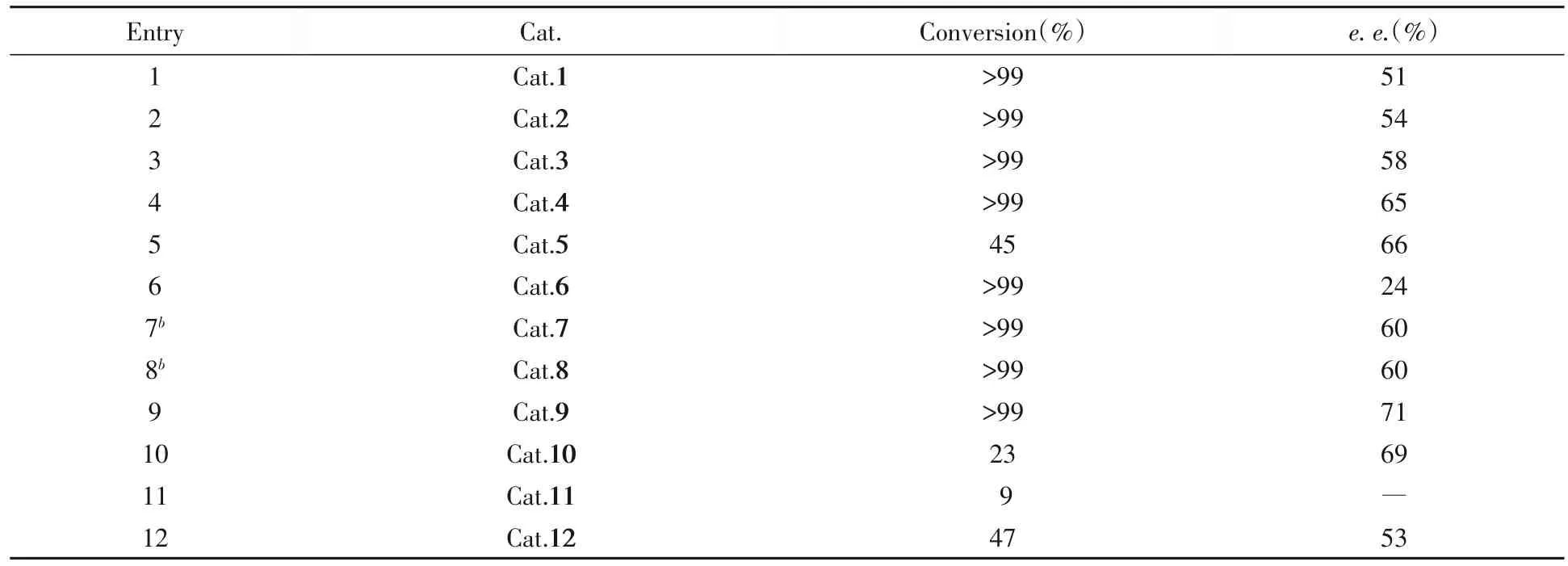
Table 4 Optimizationof catalystsa
由表4可知,吡啶环上的取代基及取代基位置对催化结果有较大的影响.在3位增加1个溴原子(表4中Entry 2),轻微地提高了对映选择性,保持同样的催化活性;在5号位引入小的供电子取代基(表4中Entries 3,4和9)能保持较好的反应活性和较好的对映选择性.其中,采用5位溴原子取代的催化剂(表4中Entry 9)获得了最高的对映选择性(71%e.e.).
除了共轭的苯基,大位阻的取代基会导致反应的活性降低(表4中Entry 6).对于5位溴原子吡啶环,采用苯丙氨酸衍生的氨基膦,可保持相似的对映选择性,但催化活性显著降低(表4中Entry 10);在6位引入取代基,反应几乎不能进行(表4中Entry 11);3,4位引入稠环结构,也显著降低反应活性(表4中Entry 12).
2.3 反应底物的考察
在确定的最佳反应条件下,对反应底物进行了拓展,结果列于表5.由表5可知,对于含有给电子取代基的底物,反应能保持较好的催化活性;对于部分缺电子取代基或稠环化合物,提高反应温度及延长反应时间可以实现全部的转化.尽管所有拓展的底物都得到较高的收率,但对映选择性不高,e.e.值处于5%~71%之间.这可能由于催化剂手性中心远离金属及本身对底物手性识别能力有限,导致底物的2种构型在氢化还原过程中转化速度差值较小.
综上所述,合成了一系列手性钌螯合催化剂,对其进行了表征,并通过单晶衍射确定了其结构.同时,通过氢化还原酯为醇的方式,尝试了α-羟基酯类化合物的动态动力学拆分,从碱的类型及其用量、反应温度、反应时间及含有不同取代基的催化剂等方面进行了研究.结果表明,基于5位溴原子的催化剂能获得较好的催化结果,在底物的拓展中表现出较好的催化活性,但仅取得中等强度的对映选择性.该反应的实现为后期进一步优化催化剂的结构并开展相关的化学研究提供了重要依据.
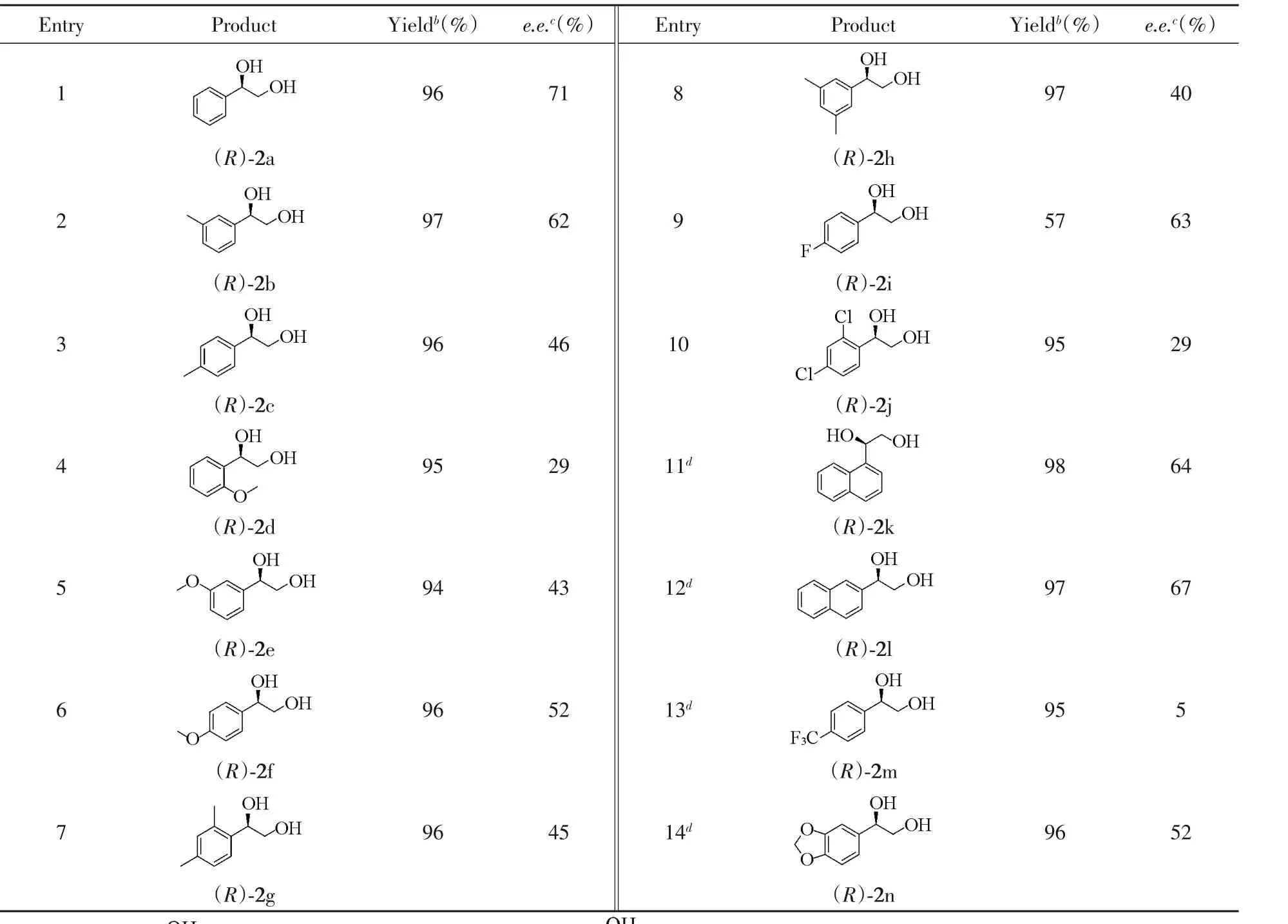
Table 5 Investigation of substrates rangea