HPLC法测定银屑舒凝胶中间体中蒽醌成分的含量
2019-09-06康宝玲李明花秦文杰裴妙荣杨晓宁梁娟娟
康宝玲 ,李明花 ,秦文杰 ,裴妙荣 ,杨晓宁 ,梁娟娟 ,李 坤
(1.山西中医药大学,山西晋中030619; 2.北京振东光明药物研究院,北京100085; 3.山西省中医药研究院,山西太原030012)
银屑舒凝胶是由山西振东制药股份有限公司研制的中药六类新药,由土大黄、苦参等药味组成,具有清热解毒、燥湿止痒之功效,用于治疗寻常型银屑病。方中土大黄为蓼科酸模属植物皱叶酸模(Rumex crispus L.)巴天酸模(Rumex patientia L.)的干燥根及根茎,常在民间使用[1],具有止血、杀虫、清热解毒和消肿的功能。文献研究表明,土大黄的主要成分有蒽醌、萘及萘醌、黄酮类及鞣质类等[2],其中大黄素有明显的体外抗菌活性[3],巴天酸模根的乙醇提取物具有较强的体外抗氧化能力,这均是其治疗银屑病的可能作用机制[4]。张燕等[5]在探索土大黄提取物治疗银屑病作用机制时发现其主要药效成分可能为蒽醌类成分,现代药理学研究表明蒽醌类成分对外周血T淋巴细胞增殖反应有抑制作用,是治疗银屑病的药效物质基础之一[6]。
在中药新药研究过程中,从药材到提取物中间体再到成药制剂,影响因素众多且环节复杂,因此,明确制备过程中的活性成分转移以及含量限定指标是中成药标准化体系建立的关键步骤。银屑舒凝胶作为企业自主研发的新药,其质量控制是前期药学研究的重要基础性工作,基于此,本文选取了蒽醌类成分中的大黄素、大黄酚和大黄素甲醚作为指标性成分,建立了同时测定银屑舒凝胶中间体中三种有效成分含量的高效液相色谱法,并对3批样品进行含量测定,实现了检验过程的快速、稳定及可控。本研究为银屑舒凝胶生产全过程的质量控制和标准体系的建立提供数据基础,具有重要意义。
1 仪器与材料
1.1 仪器
Waters2695高效液相色谱仪,Waters2998紫外检测器,Empower色谱工作站;ME204分析天平(METTLE TOLEDO);水浴锅:HH-6D1常州美特仪器制造有限公司,数控超声波清洗器(KQ-500DE型):昆山市超声仪器有限公司;Drict-Q5超纯水机。
1.2 材料
大黄素(批号:110756-200110)大黄酚(批号:110796-201379),大黄素甲醚(批号:110758-201013)以上对照品均购自中检所。处方中土大黄采挖于北京延庆和山西平顺,并经郭宝林研究员鉴定为巴天酸模,符合《北京市中药材标准》1998年版“土大黄”项下规定;苦参来自于山西振东道地药材公司,符合2015年版《中国药典》项下规定;甲醇、乙腈均为色谱纯,购自德国默克;水为超纯水;其余所用化学试剂均为分析纯。
2 方法与结果
2.1 色谱条件
phenomenex Luna C18(5 μm,4.6×250 mm),流动相为甲醇(81%)-0.1%磷酸水(19%),检测波长为254 nm,流速为1.0 mL/min,柱温为25℃。
2.2 对照品溶液的配制
精密称定大黄素对照品10.22 mg,加甲醇溶解并稀释至刻度,制得大黄素储备液(0.102 2 mg/mL);精密称定大黄酚对照品12.45 mg,加甲醇溶解并稀释至刻度,制得大黄酚储备液(0.124 5 mg/mL);精密称定大黄素甲醚对照品11.84 mg,加适量二氯甲烷溶解,并用甲醇稀释至刻度,制得大黄素甲醚储备液(0.118 4 mg/mL),3个储备液均至4℃冰箱储存。
2.3 供试品的制备
2.3.1 提取方式的考察[7]取中间体0.2 g,精密称定4份,置锥形瓶中,精密加入2.5 mol/L的硫酸20 mL、氯仿25 mL,称定质量,1、2#超声处理(功率 200 W,频率 40 kHz)1 h,3、4#加热回流(80 ℃)1 h,放冷,补重,精密量取氯仿液10 mL,蒸干,残渣加甲醇溶解,转移至10 mL的容量瓶中,摇匀,滤过,取续滤液1 mL,即得供试品溶液,按“2.1项”下的色谱条件测定,结果见表1。用水浴回流提取所测得干膏粉中总蒽醌的含量稍高于超声提取所得的含量(回流比超声所得总蒽醌的含量高4.24%),且平行性均较好,因此选择回流方式进行提取。

表1 不同提取方法所得总蒽醌含量的比较
2.3.2 酸种类的考察 依照参考文献[8]分别选用2.5 mol/L的硫酸、8%的盐酸、冰醋酸-25%盐酸(9∶1),取中间体0.2 g,精密称定6份,置锥形瓶中,1、2#精密加入2.5 mol/L的硫酸20 mL,3、4#加入8%的盐酸 20 mL,5、6# 加入冰醋酸-25%盐酸(9∶1)20 mL,同“2.3.1项”下回流方式制得供试品溶液,按“2.1项”下的色谱条件测定,结果见表2。2.5 mol/L的硫酸水解蒽醌所得含量最高为8.188 mg/g,相比8%盐酸水解蒽醌所得的含量高64.09%,比冰醋酸:25%盐酸(9∶1)水解蒽醌得的含量高58.18%,因此选择2.5 mol/L的硫酸水解蒽醌。
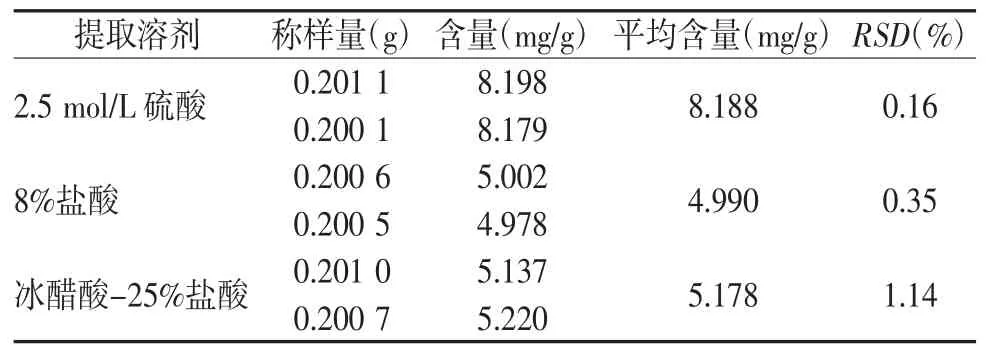
表2 不同酸种类水解所得蒽醌含量的比较
2.3.3 酸用量的考察[9]取中间体0.2 g,精密称定6份,置锥形瓶中,加入氯仿25 mL,分别精密加入 2.5 mol/L 硫酸 10、20、30 mL,称定重量,同“2.3.1项”下回流方式制得供试品溶液,按“2.1项”下的色谱条件测定,结果见表3,当酸用量随之增大时,水解得到的蒽醌的含量无明显变化,3个不同用量下的蒽醌含量的RSD值为0.86%,因此为了节省溶剂,最终选择10 mL的硫酸水解蒽醌。
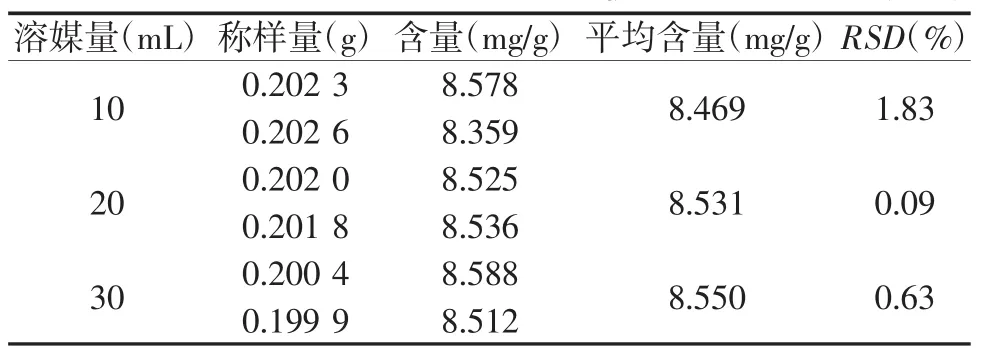
表3 不同酸用量比较 (n=2)
2.3.4 萃取量的考察 取中间体0.2 g,精密称定6份,置锥形瓶中,加2.5 mol/L的硫酸10 mL,分别精密加入氯仿 15、25、30 mL,称定重量,同“2.3.1项”下回流方式制得供试品溶液,按“2.1项”下的色谱条件测定,结果见表4,15 mL、25 mL、35 mL的氯仿量对蒽醌含量的影响不大,其RSD为0.85%,因此选择15 mL的氯仿用量进行萃取。
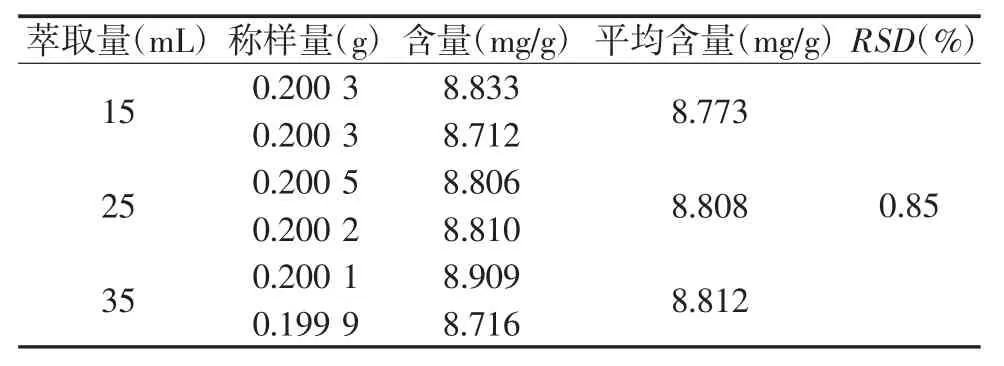
表4 不同萃取量比较
2.3.5 提取时间的考察 取中间体0.2 g,精密称定6份,置锥形瓶中,精密加入15 mL氯仿,2.5 mol/L硫酸10 mL,同“2.3.1项”下回流方式制得供试品溶液,按“2.1项”下的色谱条件测定,其中1、2#回流 60 min,3、4# 回流 90 min,5、6# 回流 120 min,结果见表5,提取60、90、120 min蒽醌含量无明显变化,其RSD为1.38%,可认为60 min已经将蒽醌类成分几乎提取完全,为节约时间,故选择最佳的提取时间为60 min。
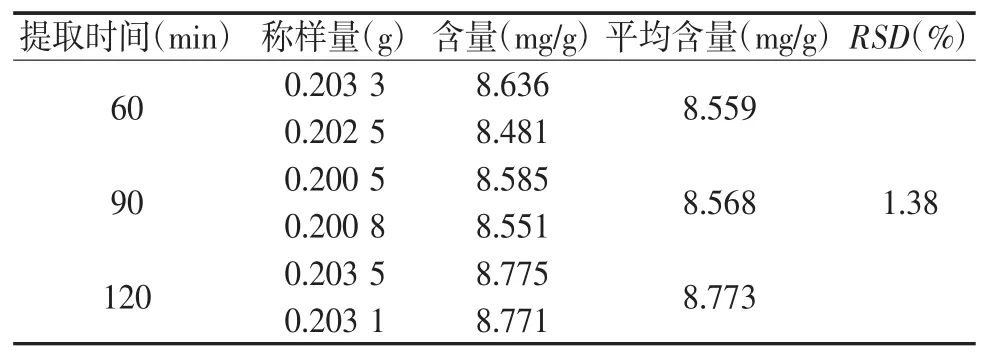
表5 不同提取时间比较
2.3.6 萃取次数的考察 取中间体0.2 g,精密称定2份,置锥形瓶中,精密加入15 mL氯仿、2.5 mol/L硫酸10 mL,称定重量,水浴回流1 h,放冷,补重,摇匀,精密量取氯仿液10 mL,蒸干,残渣加甲醇溶解,转移至10 mL的容量瓶中,取续滤液1 mL,得供试品溶液 1、2;同法操作两次,得供试品 3、4、5、6,按2.1项下测定,结果见表6,当萃取两次后蒽醌含量达到99.59%左右,在误差允许的范围内可认为已经将蒽醌萃取完全,故综合考虑选择萃取两次。
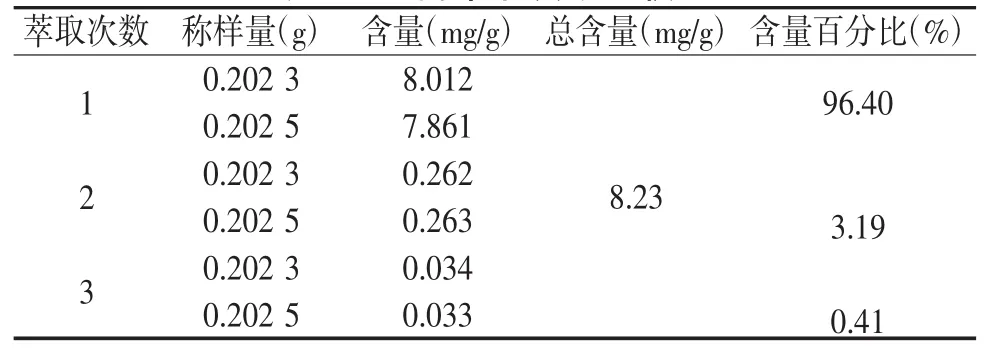
表6 不同萃取次数比较
2.3.7 供试品溶液制备的最优工艺 取中间体0.2 g置锥形瓶中,精密称定,加2.5 mol/L的硫酸溶液10 mL、氯仿15 mL,称定重量,加热回流(80℃)1 h,放冷,补重,精密量取氯仿液10 mL于蒸发皿中,同法操作两次,合并两次萃取液,蒸干,残渣加甲醇溶解,并转移至10 mL的容量瓶中,摇匀取续滤液即得。
2.4 方法学考察
2.4.1 线性关系的考察 分别精密吸取“2.2项”下的大黄素、大黄酚、大黄素甲醚储备液适量,加甲醇稀释制得质量浓度分别为5.11、10.22、25.55、51.1、102.2 μg/mL的大黄素对照品溶液,质量浓度分别为 6.22、12.45、24.9、62.25、124.5 μg/mL 的大黄酚对照品溶液,质量浓度分别为5.92、11.84、23.68、59.2、118.4 μg/mL 的大黄素甲醚对照品溶液。按“2.1项”下的测定,得标准曲线方程,结果见表7,式中X代表浓度,Y代表峰面积。
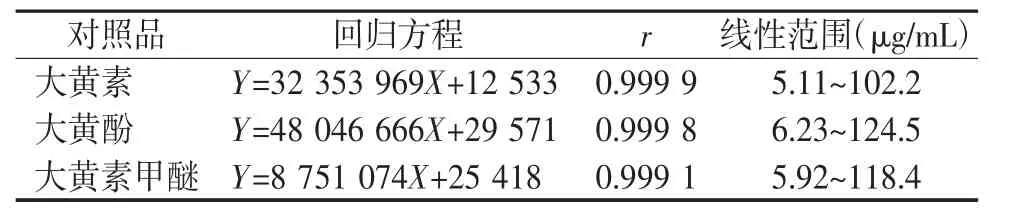
表7 3个成分的线性回归方程
2.4.2 专属性考察 按处方比例及工艺路线制成缺土大黄的阴性中间体,再按照“2.3.7项”下方法制得阴性供试品样品,按“2.1项”下测定,结果发现,阴性供试品样品溶液色谱图在大黄素、大黄酚和大黄素甲醚处无吸收,说明阴性样品无干扰,结果见图1。
2.4.3 重复性考察 取同一批中间体0.2 g,精密称定6份,按“2.3.7项”下平行制备供试品溶液,在“2.1项”下测定并计算,结果表明大黄素、大黄酚、大黄素甲醚的平均含量分别为3.332、3.343、0.973 mg/g,RSD为1.21%、0.73%、1.93%,表明该方法重复性良好。
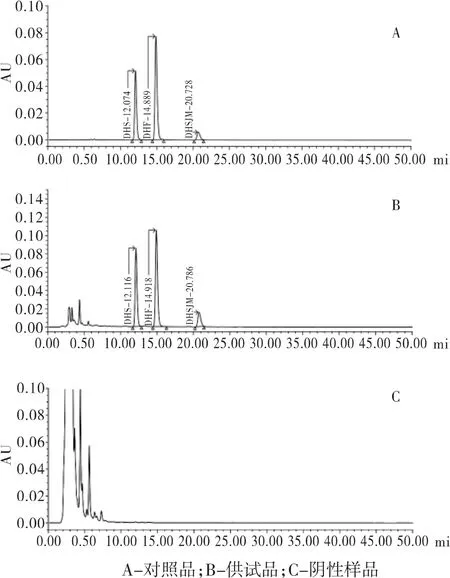
图1 蒽醌含量测定专属性考察HPLC图
2.4.4 中间精密度考察 让实验室两名实验员按“2.3.7项”下平行制备供试品溶液6份,在“2.1项”下进行测定并计算,结果不同实验员用不同的仪器测得的大黄素、大黄酚、大黄素甲醚的含量的RSD分别为1.17%、0.83%、2.42%,可认为该样品制备方法的中间精密度良好。
2.4.5 稳定性试验 取同一供试品溶液,按“2.1项”下分别在 0、2、4、8、12、24 h 进样测定,结果大黄素、大黄酚、大黄素甲醚含量的RSD分别为0.49%、0.46%、0.40%,表明供试品溶液在24 h内稳定。
2.4.6 加样回收率试验 精密称定中间体0.1 g,分别按表8要求加入对照品溶液,按“2.3.7项”下方法制备,在“2.1项”下测定并计算。结果见表8。
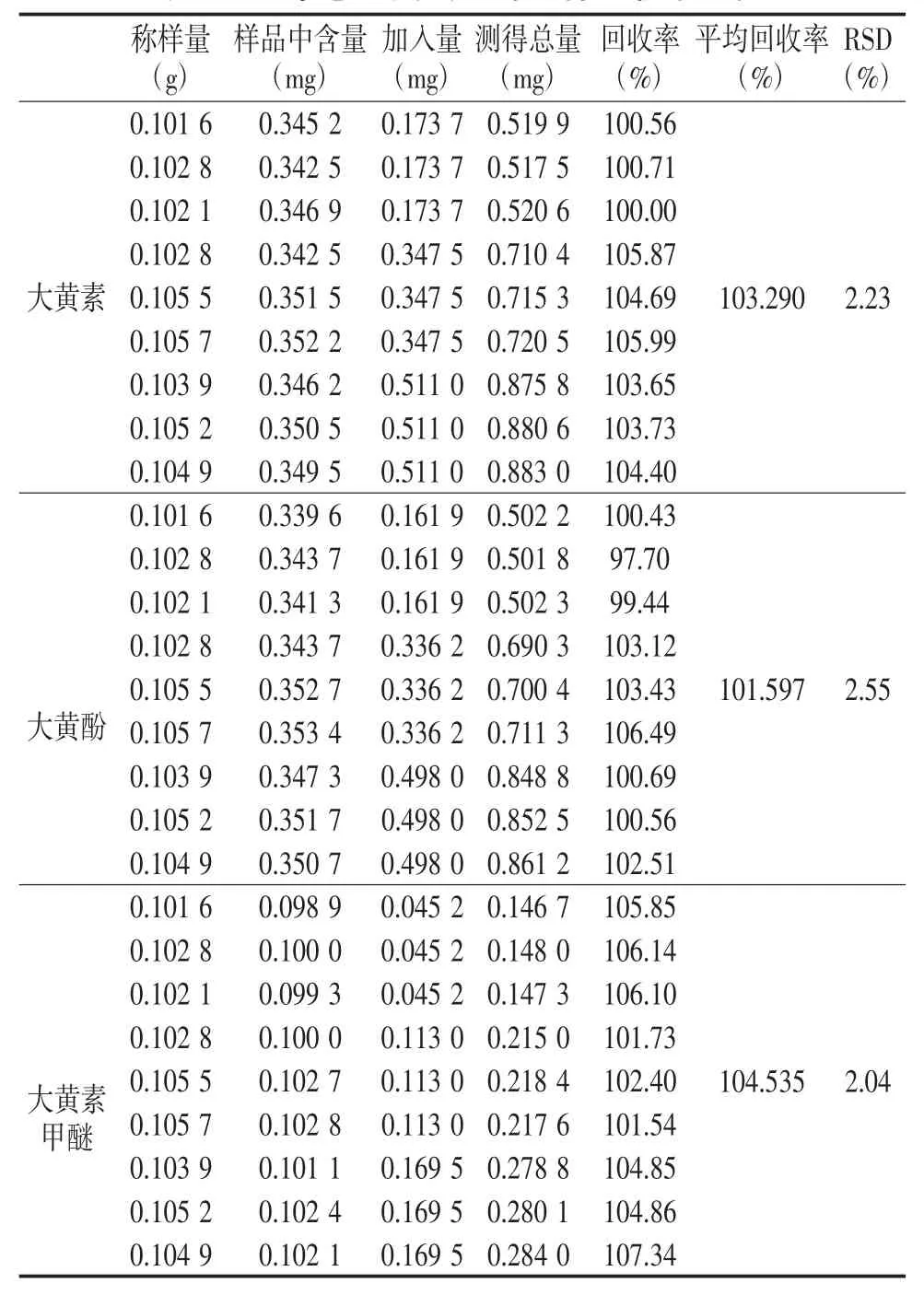
表8 三个蒽醌类成分的加样回收率试验
2.5 样品含量测定
分别取3批样品,按照“2.3.7项”下方法制备,在“2.1项”下测定,结果中间体中大黄素、大黄酚、大黄素甲醚的含量分别为3.575、3.531、1.039 8 mg/g。
3 讨论
在试验中分别考察不同流速(0.9 mL/min,1.0 mL/min,1.1 mL/min)、不同波长(250 nm,254 nm,258 nm)、不同温度(20℃,25℃,30℃)、不同流动相比例[甲醇-0.1%磷酸水=(79∶21,81∶19,83∶17)、及不同厂家的 C18色谱柱(Phenomenex,Agilent,Waters bridge]。结果随着增大流速或流动相比例时,3个成分的出峰时间相对早,但分离度均>6.0,对称因子在1.01~1.08,均符合要求;当改变柱温和波长时,其出峰时间、分离度、对称因子均没有变化,且含量的RSD分别为0.97%和0.45%;不同色谱柱考察结果表明蒽醌的分离对色谱柱的选择性不强,3个色谱柱均可以使蒽醌的峰离开,且分离度、校正因子均符合要求,所测得的含量的RSD为1.10%,故该检测方法的耐用性良好。
本文中所建立的高效液相色谱条件过程简单、快速,方法学考察符合要求,稳定性高、重复性好、准确度高,可用于银屑舒凝胶中间体中指标性成分的测定,为中间体及成型凝胶制剂的质量控制提供切实可行的检测方法,为后续产品质量标准的制订提供了数据支撑。
