局部晚期直肠癌新辅助化放疗病理学缓解的预测基因分析
2019-07-02代佳佳李松霖
代佳佳,肖 何,张 琴,李松霖,陈 川,王 阁
0 引 言
对于局部晚期直肠癌(locally advanced rectal cancer,LARC)患者,术前新辅助化放疗(neoadjuvant radiochemotherapy,NCRT)后进行全直肠系膜切除术(total mesorectal excision,TME)是目前的标准治疗方式[1-3]。相对于术后辅助放化疗,NCRT可降低术前分期、提高手术切除率和保肛率,并显著降低局部复发率[4-5]。然而在临床实践中,仅10%~30%的患者显示病理完全缓解[6-8]。因此除术后TNM分期以外,术后病理结果显示的肿瘤退缩分级(tumor regression grading,TRG)也NCRT效果评价的一项重要参考指标。
美国国立综合癌症网络(National Comprehensice Cancer Netword,NCCN)指南推荐评估肿瘤治疗反应的病理学分级系统包括完全反应(TRG4):无活的癌细胞残留;中度反应(TRG3):单个或小簇癌细胞残留;轻度反应(TRG2):残留癌灶;反应不良(TRG1):仅少数或未见癌细胞消退[7]。研究表明,术前NCRT后肿瘤完全退缩(TRG4)和部分退缩(TRG3)与患者无复发生存和总生存密切相关,肿瘤达到完全退缩的患者较退缩情况较差患者具有明显的生存获益[9-10]。目前,第7版美国癌症联合委员会(American Joint Commettee on Cancer,AJCC)分期已提出对直肠癌标本检查时应评价新辅助治疗后的治疗反应[11]。因此,对于术前放化疗后能到达TRG 3/4级人群的预测有助于筛选出对NCRT最大获益人群和避免对放化疗不敏感患者的毒副反应,对促进直肠癌的个体化治疗有重要的临床意义。
迄今已有大量文献报道了各种临床和分子生物学指标对NCRT完全病理学缓解的影响因素及预测模型的构建[12-18]。其中包括治疗前肿瘤大小和肠系膜淋巴结转移状态等临床特征[12]、核磁共振影像组学特征[13]、肿瘤芽殖等病理形态特征[14]以及放化疗敏感性相关基因标签等[15-18];而基于基因标签的预测模型除具有潜在的临床应用价值外,还因与肿瘤放疗抵抗机制密切相关而具有放射生物学探索的价值。从样本量考虑,合并多个数据集扩大训练集样本量对于构建稳定的预测模型具有重要作用[19]。因此,本研究试图综合分析4个局部晚期直肠癌NCRT基因表达谱数据集和肿瘤退缩等级,构建预测模型以进一步判断该类模型的临床应用价值和潜在的直肠癌放化疗抵抗机制。
1 资料与方法
1.1 数据集的获取和前处理以“preoperative chemoradiotherapy”和“rectal cancer”或“neoadjuvant chemoradi otherapy”和“rectal cancer”作为检索词搜索GEO数据库,选择可获取基因微阵列表达值和肿瘤退缩分级的数据集,排除RNAseq、非编码RNA数据集和没有标示退缩等级的数据集。共有4个数据集符合入组标准,数据集名称和相关信息见表1。采用R软件包“annotate”将各微阵列探针ID号注释到基因符号。对于多个探针对应1个基因,取4分位距最大探针代表该基因表达值。以基因符号合并 GSE35452、GSE46862、GSE68204 3个数据集共174个样本,其中79例达到TRG 3/4,缓解率为44.1%。利用“virtualArray”包函数“virtualArrayCom-Bat”对合并数据集的批次效应进行校正[20]。δ统计量和主成分分析判断批次效应校正结果[21]。
1.2 基于LASSO的Logistic回归和SVM法筛选候选基因和模型构建R软件包“sample”函数随机将174例合并数据集按7∶3比例分成训练集(n=121)和验证集(n=53)。对训练集采用单因素Logistic回归筛选与TRG 3/4显著相关的基因,依据回归系数显著性P值对基因进行排秩,P<0.05作为纳入最小绝对值收敛和选择算子(least absolute shrinkage and selection operator,LOSSA)算法的候选基因。LASSO算法构建预测模型,取λ对应内部交叉验证错配率最小值对应变量作为模型变量,并在验证集中判断该模型对TRG 3/4的诊断准确性、特异性和灵敏度。在训练集中对每个基因利用t检验分析TRG3/4与TRG0/1/2组间表达值差异,并根据显著性P值对基因进行排秩,取前50个基因利用支持向量机(support vector machine,SVM)构建预测模型,在验证集中判断该模型对TRG 3/4的诊断准确性、特异性和灵敏度,SVM算法各参数使用缺失值。以上2种分析分别反复随机取样500次,以判断模型预测作用的稳定性;并对基因排秩取平均值和标准差以确定排秩前100基因的可重复性。LASSO算法和SVM分别采用R软件包“glmnet”和“penalizedSVM”完成。
LASSO算法纳入模型最多的前30个基因与独立验证集GSE53781有重叠的基因共21个,再次在174例合并数据集中验证该21个基因对病理学缓解预测效能。同时在GSE53781中采用这21个基因单因素Logistic回归系数与表达值乘积和作为化放疗敏感指数(sensitivity index,SI)以判断该指数对病理学缓解的预测作用。
1.3 基于差异表达基因和转录调控网络分析和基因功能注释在174例合并数据集中。利用“limma”包分析TRG3/4与TRG0/1/2组间差异表达基因。将多重校正前P<0.05基因纳入转录网络调控分析,判断差异表达基因潜在调控机制[22]。“reactome”包用于候选基因的功能注释。
2 结 果
2.1 在 GSE35452、GSE46862、GSE68204构成的数据集中构建和验证预测模型合并3个数据集后共12 803个基因纳入分析。批次效应校正前后δ统计量分别为2.350(P=0.008)和0.0257(P=1.000),主成分分析亦表明批次效应校正后3个数据集来源病例在前2个主成分中无聚集性分布,见图1。单因素Logistic回归表明在500次随机抽样生成的训练集中,回归系数显著性P值最小的前100个基因P值均较大,仅3个基因P<0.01,且有较大的变异幅度,见图2。同样,基因排秩靠前的100个基因平均秩也较大,并存在较大的变异幅度。LOSSA模型在训练集中的诊断准确性、特异性和灵敏度分别为0.998(95%CI:0.991~1.000)、1.000(95%CI:1.000~1.000)、0.998(95%CI:0.981~1.000)。但在验证集中却得到较弱的诊断效能,准确性、特异性和灵敏度分别为 0.523(95%CI:0.396~0.642)、0.578(95%CI:0.373~0.762)、0.464(95%CI:0.258~0.700)。同样,SVM法在验证集中的准确性、特异性和灵敏度分别为 0.504(95%CI:0.377~0.623)、0.596(95%CI:0.393~0.830)、0.405(95%CI:0.182~0.650)。见图3。

表1 所用数据库名称和微阵列芯片Table 1 Series of GEO accession and platforms used for the analysis
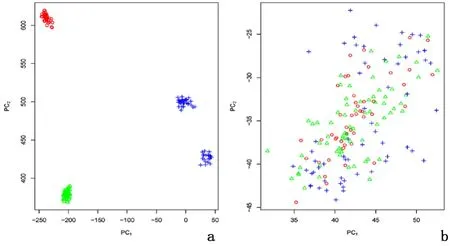
图1 GSE35452、GSE46862、GSE68204数据集合并后批次效应校正前后主成分分析Figure 1 Principal component analysis before and after batch effect correction after merging three data sets
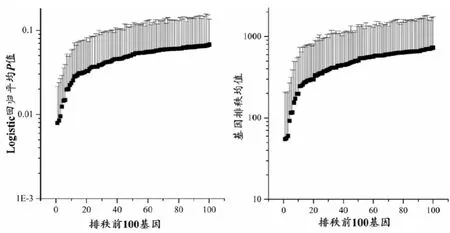
图2 单因素Logistic回归系数显著性最高的前100个基因平均P值及排秩Figure 2 Average P values and ranks of the first 100 genes with the highest significance of coefficients from univariate Logistic regression in the training set
2.2 GSE53781作为外部验证集验证候选基因在500次训练集中引入LASSO算法模型次数最多的前30个基因中,有21个与GSE53781重叠。取该21个基因作为独立验证的候选基因。LASSO算法表明这21个基因在合并数据集中对病理学完全缓解和接近病理学完全缓解病例的诊断准确性、特异性和灵敏度分别为 0.816、0.842、0.785。其调谐参数λ与对应的错配率见图4。敏感指标病理学缓解诊断的AUC为0.863(95%CI:0.811~0.912)。21个候选基因符号和功能注释以及回归系数见表2。GSE53781数据集中有26例,其中10例达到病理学缓解,该组人群中计算得到的敏感指标1.674~54.985,中位值为7.133)。敏感指标有较高诊断效能(AUC=0.925,95%CI:0.817~1.000),见图4。但是,对GSE53781随机取出21个基因重复分析1000次得到对病理学缓解诊断的AUC 95%CI:0.769~0.969,21基因对应的AUC处于该CI内。
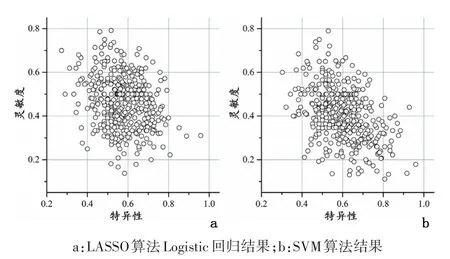
图3 不同方法对新辅助化放疗缓解诊断的特异性和灵敏度散点图Figure 3 Scatter plots showing the sensitivity and specificity of the predictive model for response to neoadjuvant chemoradiotherapy in validation sets in 500 times of random sampling
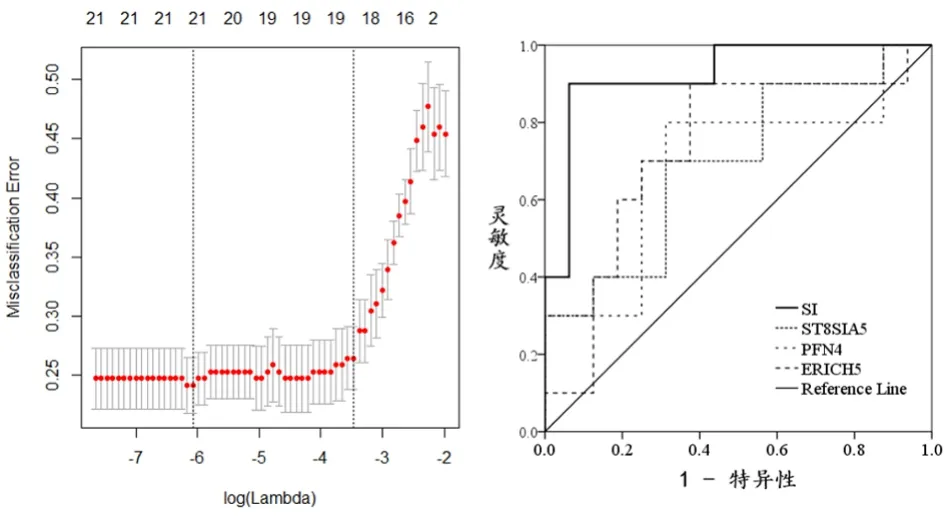
图4 合并数据集中21基因LASSO算法λ与内部交叉验证错配率关系图Figure 4 Relationship between internal cross-validation mismatch rate and lambda in LASSO algorithm with 21 candidate genes in the 174 merged datasets
2.3 LARC化放疗抵抗基因参与调控网络在174例合并数据集12803个基因中,差异表达基因共505个。RNEA分析表明,这些差异表达基因主要受 NF-κB、CTNNB1、IL6/STAT3、E2F1、GLI2 以及MYC转录因子调控基因,见图5。
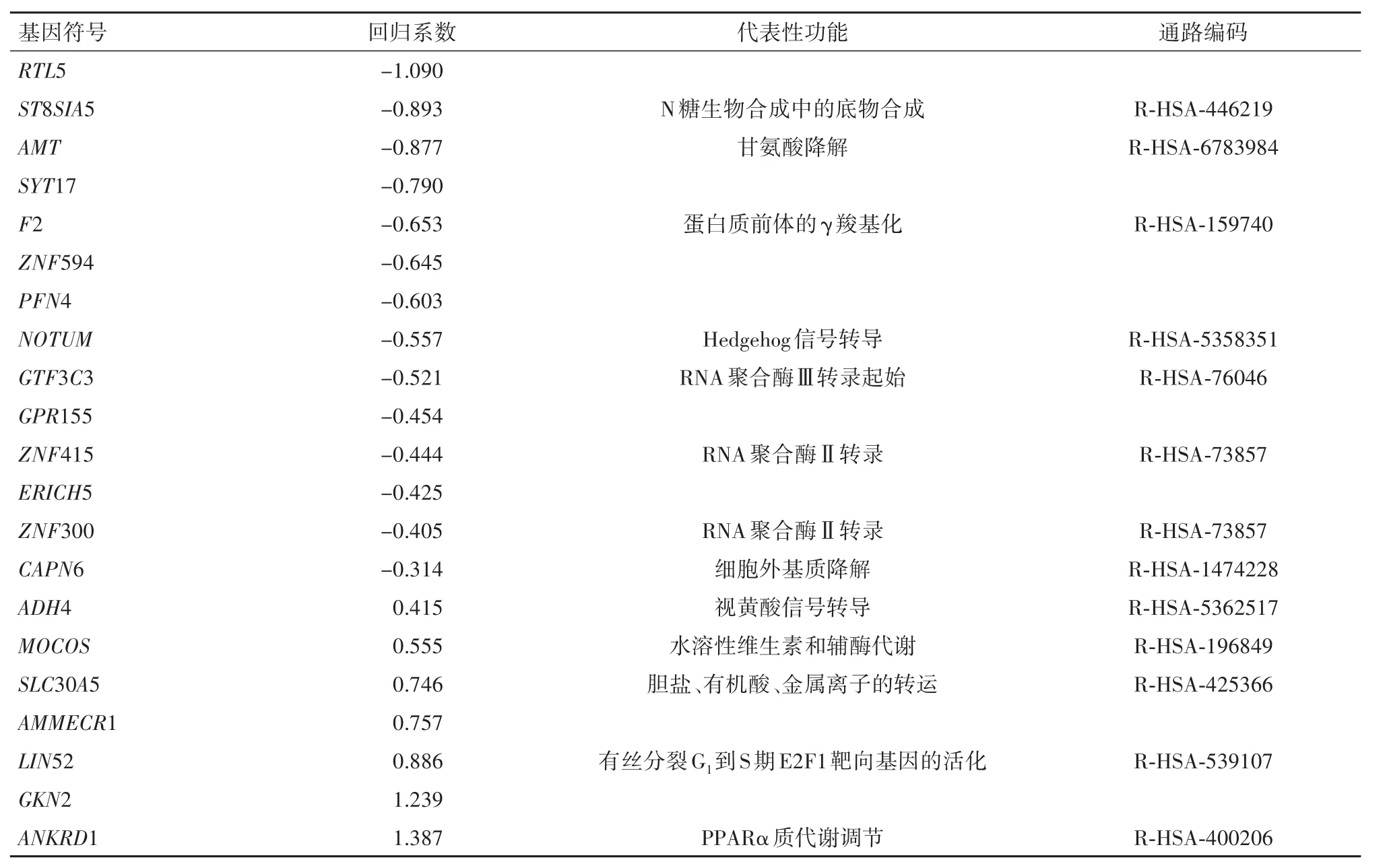
表2 21个候选基因在合并数据集中Logistic回归系数和部分功能注释Table 2 Logistic regression coefficients and functional annotations of 21 candidate genes in the 174 merged datasets
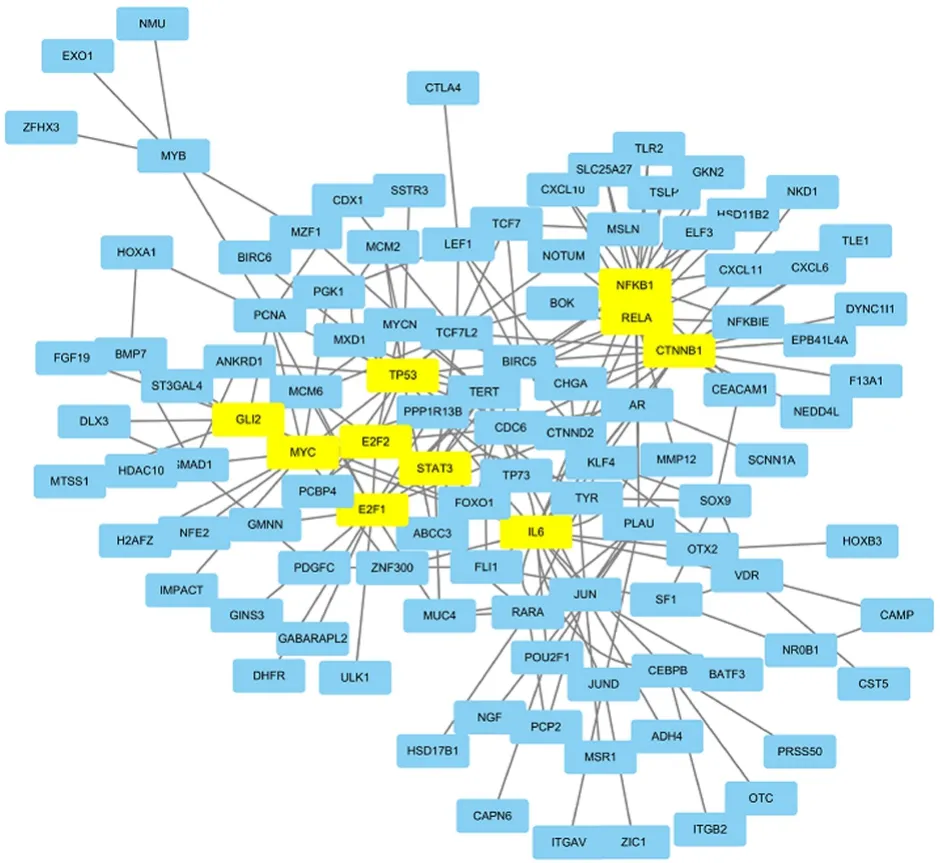
图 5 GSE35452、GSE46862、GSE68204 3个合并数据集中RNEA揭示缓解组与未缓解组间505个差异表达基因转录调控网络图Figure 5 Transcription regulatory network of the 505 differentially expressed genes in the response and non-response groups in the 174 merged datasets
3 讨 论
目前已有大量基于高通量基因组学和生物信息学分析的肿瘤放射内在敏感性研究试图建立个体化放疗指标[23]。对于局部晚期直肠癌,已有若干研究筛选出了可构建有效的NCRT病理学缓解或完全缓解预测模型的候选基因[15-18]。但这些基于统计量排秩而选择“重要”基因纳入模型构建的方法易受训练集样本量的影响。足够样本量的训练集才能得到稳定的标签基因和预测效能。合并多个数据集的综合分析可能具有提高构建预测模型稳定性的优势[19,24]。本研究合并了 GEO 数据库中的GSE35452、GSE46862、GSE68204 3个数据集,共纳入了12 803个基因数据值,通过在缓解组和未缓解组间采用Logistic回归和t检验,得出了平均排秩最小的100个基因,但这100个基因在121例训练集中并不稳定;而且在不同次取样构成的训练集中,同一基因的排秩存在较大偏差,结合在各次取样中对53例内部验证集较弱的预测效能。该结果提示预测模型具有一定的预测能力,但验证效能不足,尚需进一步深入研究。有文献报道其他二分类临床结局预测模型仅使用了 120 例样本已足够[19,24];但本研究单次用174例作为训练集也不足以得到稳定的标签基因,且在不同的研究中得到的标签基因种类很少重叠[15-18]。提示了参与直肠癌NRCT抵抗或敏感基因种类在人群间存在较大异质性,其机制仍需进一步探讨。研究表明,在结直肠癌分子分型基础上进行预后标志物筛选,或者联合肿瘤细胞内在基因与肿瘤微环境组成细胞如肿瘤相关成纤维细胞、各种肿瘤浸润免疫细胞亚群基因表达构建疗效预测模型是克服肿瘤异质性的有效手段[25-26]。
本研究筛选出的21个预测基因可能与直肠癌放疗和化疗敏感性有密切联系,涉及糖代谢、丝氨酸/甘氨酸合成与代谢、促凝血机制、转录调控和细胞周期调控等。如ST8SIA5基因属于糖基转移酶家族29,参与合成多种神经节糖苷。同属一个家族的另一个酶ST8SIA1则具有维持三阴乳腺癌细胞干性特征,并通过FAK-AKT-mTOR促进转移的作用[27]。AMT基因编码氨基甲基转移酶是构成甘氨酸解理系统蛋白之一。线粒体丝氨酸/甘氨酸代谢系统对快速增殖肿瘤细胞所需的嘌呤核苷合成具有重要作用。而甘氨酸解理系统可清除该过程中产生的过量甘氨酸,否则会在胞内会产生氨基酮和丙酮醛等细胞毒性物质[28]。而甘氨酸自身也可通过其受体抑制内皮细胞血管生成和结直肠癌细胞转移[29]。快速增殖的肿瘤细胞很容易造成乏氧和瘤体中心区域的坏死。推测AMT可能对于维持直肠癌在快速增殖情况下改善乏氧并诱导肿瘤转移有重要作用。因此,本研究结果提示AMT基因表达越高的直肠癌患者达到病理学缓解的概率越低。
此外,本研究发现凝血酶(F2)表达可能对放疗抵抗有重要影响。凝血酶介导的纤维蛋白/血小板凝聚被认为是肿瘤细胞微转移灶形成的机制之一。凝聚的血小板通过TGF-β1减弱自然杀伤细胞的功能,使微转移肿瘤细胞逃避免疫监视[30]。大量研究提示,上皮间质转化或干性转化的肿瘤细胞对放疗和化疗药物具有较强的抵抗作用[31-32]。而本研究中,对缓解与未缓解组间差异表达基因转录调控网络分析也提示这些差异表达基因主要受IL-6/STAT3、NF-kB和MYC等与肿瘤转移或干性表型等相关转录因子调控。如IL-6不但可促进结直肠癌细胞系HCT116和HT29上皮间叶转化和血管拟态的形成,且与转移密切相关,同时还可增强A549和H157非小细胞肺癌细胞系CD133+干细胞对放疗诱导双链断裂的修复能力和抵抗凋亡的能力[31-32]。临床研究也表明,浸润能力和淋巴结转移也是直肠癌病理学完全缓解的不良预测因素[12,14]。因此,结合既往报道可以推测直肠癌中参与上皮间质转化和干性转化机制并促进转移的肿瘤微环境就是参与放化疗抵抗的重要机制。
本研究纳入分析的4个GEO数据集采用了不同的病理学缓解分级标准、所用杂交芯片检测平台不尽相同,微整列芯片重叠基因存在有限性以及未分细胞亚群分析等因素均成为本研究的局限性。尽管本研究并未建立明确的预测模型,分析结果提示直肠癌参与放化疗抵抗的机制可能与其浸润、转移机制相关,并且强调了参与放化疗抵抗基因在人群间的异质性和基于肿瘤及其微环境多种细胞亚群研究的重要性。
