青海汉族牙周病患者口腔微生物的群落结构分析
2018-12-12李志艳赵翀刘静张欣朱德锐陈筠
李志艳 赵翀 刘静 张欣 朱德锐 陈筠
研究发现,菌斑微生物是牙周炎发生的起始因子,受到宿主的遗传背景、饮食、生活环境及文化背景等因素的制约影响[1-2]。牙周病可疑致病菌主要有福赛斯坦纳菌、伴放线菌嗜血菌、牙龈卟啉单胞菌、具核梭杆菌、中间普氏菌、齿垢密螺旋体等[3-4]。青海地区地处高原,多民族聚居,汉族人群约占53.02%,高原缺氧等因素会对厌氧菌和牙周组织均造成影响,且牙周病患病率较其他平原地区高[5-6]。目前,有关口腔微生物的研究采用高通量测序技术(high throughput sequencing)是国内外研究热点[7-8],相关研究在青海地区研究报道甚少。本研究采用16S rRNA基因高通量测序技术探究青海汉族牙周病患者口腔微生物的多样性和群落结构,为后期口腔疾病的预防、诊断与治疗提供理论依据和技术支持。
1 对象与方法
1.1 研究对象
病例选自青海大学附属医院口腔内科门诊慢性牙周炎患者10 例、正常人群(对照组)5 例唾液样本共15 例,其中长期在青海居住10 年以上,男7 人,女8 人,年龄范围39~66 岁以上,平均年龄(45.6±2.54) 岁。其中慢性牙周炎患者 (根据第三次全国口腔流行病学调查标准[9]),纳入标准:①牙周袋深度(PD)≥4 mm或附着丧失量(AL)≥2 mm;② X线片示牙槽骨吸收超过根长的1/3。所有选取的对象无糖尿病、心血管病、甲亢等系统性疾病。过去3 个月内未使用过抗生素和消炎药物等,就诊前6 个月内未做口腔治疗,并排除侵袭性牙周炎。
1.2 样本采集
唾液样本的收集:清晨起床后,刷牙洗漱之前或两餐之间,于10 点钟左右或16 点钟左右。采集样本前让受检者用温开水轻轻漱去口中的食物残渣,然后采集口腔自然排出的唾液于无菌容器内,唾液量0.5~1.0 ml。盖上管口或容器盖立即送检,不能及时送检的唾液最好加盖少量无菌石蜡油以隔绝空气。
1.3 基因组DNA的提取及质量控制
样本于0.22 μm 细菌滤膜上进行真空抽滤,将滤膜剪碎放入DNA 试剂盒(QI Aamp Fast DNA Stool Mini Kit,Qiagen,德国),参照试剂盒步骤提取基因组DNA,2.0%琼脂糖凝胶电泳分析环境样本DNA完整性。DNA质量纯度检测分析采用微量检测仪Microplate Reader(MD公司,USA),合格样品总DNA保存于-80 ℃。
1.4 PCR扩增16S rDNA和测序分析
细菌使用16S rRNA基因通用引物341F(5’-CCTACGGGNGGCWGCAG-3’)和785R(5’-GACTACHVGGGTATCTAATCC-3’)进行V3-V4目的基因扩增(ABI Gene Amp 9700, USA)。PCR反应条件为:95 ℃(5 min),94 ℃(45 s),55 ℃(30 s),72 ℃(90 s),共35 个循环,最后在72 ℃下延伸5 min。采用Axyprep DNA Gel Extraction Kit(Axygen scientific,USA)进行PCR扩增产物纯化;采用Qubit®2.0荧光计(Invitrogen, USA)进行DNA精准定量分析。检测合格的PCR纯化产物进行16S rRNA基因量测序(MiSeq/454/Sanger,3730xl,ABI,USA),由上海惠研生物科技有限公司完成,每个样本获得双端序列数据为2~3 万个reads。根据测序reads之间overlap关系进行拼接(merge),同时对reads质量和merge效果进行质控过滤(Q30>80%),根据序列首尾两端的barcode和引物序列区分,优化有效序列。
1.5 测序结果分析
使用QIIME 1.8.0软件对原始数据进行处理,在相似度97%水平上聚类出可操作分类单元OTU(Operational taxonomic unit),进行OTU picking和物种注释分析。使用alpha稀释曲线(Alpha rarefaction.py)计算了样品的菌群多样性香农指数(Shannon)、谱系多样性(PD whole tree)、观察物种(Observed species)和菌种丰富度指数chao1,以及基于OTU进行主成分分析(PCA, principal component analysis)。根据分类学分析结果(纲Classes、属Genera与种Species)分析微生物的群落结构、物种组成比例及丰度[9]。
2 结 果
2.1 高通量测序深度分析
通过Illumina MiSeq测序平台对青海牙周病患者与正常人群口腔唾液样本细菌16S rRNA基因(V3-V4区)生物多样性检测,共计获得10 587条reads,每个样本平均读取706 条reads,平均读长250 bp。随着样本测序深度的增加,稀释曲线趋向平坦,获得的OTU数目变化不大,说明测序数据量合理。在相似度97%水平上聚类OTU,并进行OTU picking,获得青海地区牙周病患者组与正常人群口腔唾液样本微生物物种注释OTU数目分别为60和14。表明青海地区牙周病患者组的总观察物种数量明显多与正常组。
2.2 群落结构及优势种群分析
通过在Ribosomal Database Project(RDP)数据库内进行相似性比对,并分别在门、纲、属的层次下对各样品物种丰度进行统计分析 (图 1)。牙周病组优势种群细菌依次为(相对丰度>1%):肠杆菌科未分类种(Enterobacteriaceaeunclassified)、假单胞菌属未知种(Pseudomonas)、不动杆菌属未知种(Acinetobacter)、蜡样芽胞杆菌(Bacilluscereus)、气单胞菌科未分类种(Aeromonadaceae)、约氏不动杆菌(Acinetobacterjohnsonii)、奈氏球菌属未知种(Neisseria)、梭杆菌属未知种(Fusobacterium)、金黄葡萄球菌(Staphylococcusaureus)、副流感嗜血杆菌(Haemophilusparainfluenzae)、乳球菌属未知种(Lactococcus)、肺嗜冷杆菌(Psychrobacterpulmonis)、Wautersiella、链球菌属未知种(Streptococcus)、鲁氏不动杆菌(Acinetobacterlwoffii)、颗粒链菌属(Granulicatella)。正常组有4 种优势菌属仅检测出4 类种群Mucilaginosasp.、Enterococcussp.、乳酸杆菌目未分类种(Lactobacillalesunclassified)和Chryseobacteriumsp.。综上分析表明,患病组的微生物种类繁多,而正常组微生物种类相对单一。牙周病组的门纲分类和群落结构相对复杂于正常组,表明牙周病发生与口腔微生物群落变化存在相关性。
2.3 微生物多样性性分析
基于OTU注释和测序统计进行微生物多样性分析(表 1),表明牙周病组检测出观察物种数量大于正常组观察物种数量,此结果与高通量测序获得的OTU丰富度Chao1指数结果相一致。Shannon指数结果分析表明牙周病组的微生物多样性显著高于正常组。根据PD whole tree指数显示,牙周病组(0.86~2.33)的微生物系统发育距离总和大于正常组(≤1.00),表明牙周病可疑致病微生物群落结构远远复杂于正常组,系统发育关系亦趋于复杂。
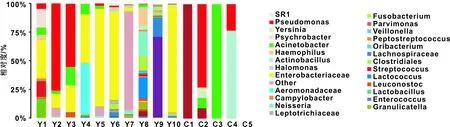
图 1 微生物种水平相对丰度统计分析牙周病患者(Y)、正常组(C)
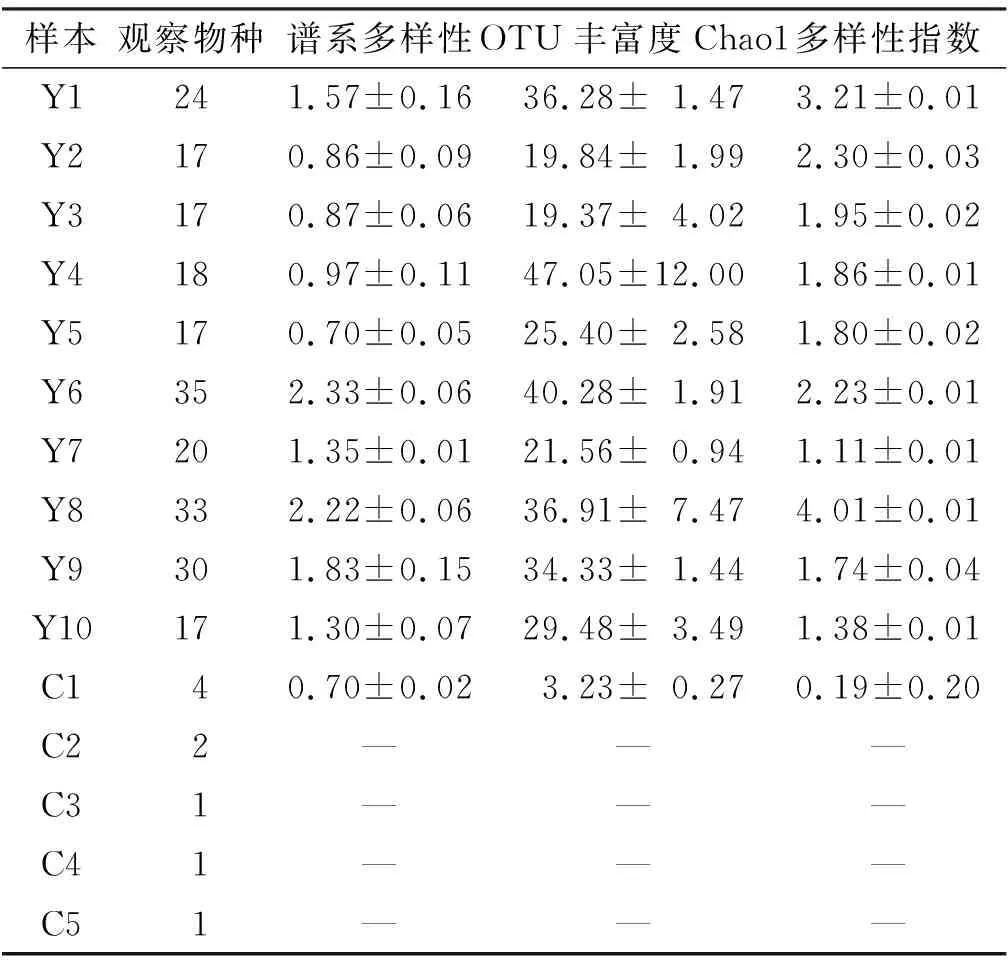
表 2 西宁市汉族人群牙周病患者(Y)和正常者(C)口腔微生物多样性统计分析
注:菌株单一,无统计意义
3 讨 论
目前,高通量Illumina MiSeq测序是微生物多样性研究的重要技术手段,测序精确,数据准确度高,可全面反应复杂样品的微生物群落的组成。本研究通过高通量测序的方法对青海汉族牙周病组(10 例)、正常组(5 例)口腔唾液样本中的微生物群落结构和多样性进行了初步研究,物种注释筛选获得青海地区牙周病组60 个OTU,共计5 门9 纲33 属;正常人群组14 个OTU,共计2 门2 纲4 属。可见牙周病组口腔微生物物种明显较正常人多,说明菌斑微生物和牙周炎的发病是密切相关的。
本研究2 组样本(15 例)的物种丰度统计分析,牙周病组检测出的微生物种类较多,包括有变形菌门(Proteobacteria,78.24%)、厚壁菌门(Firmicutes,16.33%)、梭杆菌门(Fusobacteria,2.44%)、拟杆菌门(Bacteroidetes,2.46%)。正常组的检出种类主要是Proteobacteria(检出率为26.73%)和Firmicutes(检出率为41.82%)。吴芳等[10]采用16S rRNA基因克隆文库法分析甘肃东乡族牙周炎患者和健康人(各5 例)唾液微生物多样性,检出牙周炎患者的主要类群是Firmicutes(53.80%)、Proteobacteria(23.10%)、Bacteroidetes(11.0%)和Fusobacteria(4.0%)。通过比较分析可知,口腔微生物的组成类群基本一致,微生物群落结构变化和丰度比例不尽相同,这可能与受试者居住环境、生活习惯的差异而造成,处于低压、低氧等环境因素导致口腔内大多数细菌营兼性或者专性厌氧及异养生活的变形菌门丰度比例较高,而牙周病患者口腔内,由此亦表明口腔常见疾病的发生与发展,是口腔内多种微生物共同作用的结果。
口腔微生物常以群体的方式导致各种口腔疾病,群体中的每一个个体均参与了疾病的发生发展过程,即使是低丰度的成员依然可能作为复杂的群落行为表现的关键物种[11]。牙周炎是由牙菌斑中多种微生物引发的牙周组织慢性感染性炎症疾病。研究表明,卟啉单胞菌(Porphyromonas)、齿垢密螺旋体(Treponemadenticola)、福赛斯坦纳菌(Tannerellaforsythia)、拟杆菌(Bacteroides)、隐藏真杆菌(Eubacteriumsaphenum)、消化链球菌(Peptostreptococcus)、 栖石普雷沃菌(Prevotelladenticola)、 微小微单胞菌(Parvimonasmicra)、小杆菌(Dialister)、龈沟产线菌(Filifactoralocis)、 脱硫球茎菌(Desulfobulbusspecies),互养菌和龈沟螺杆菌等与该病密切相关[12]。吴芳等[13]对照分析牙周炎患者与健康患者,其优势属群依次为链球菌(Streptococcus,40.3%)、奈瑟氏球菌(Neisseria,11.4%)、兼性双球菌(Gemella,6.02%)、韦荣球菌(Veillonella,4.22%)、聚糖杆菌(Aggregatibacter,3.16%)、卟啉单胞菌属(Porphyromonas,3.01%)、福氏杆菌(Fusobacterium,2.14%)、嗜血杆菌(Haemophilus,2.14%)、纤毛菌属(Leptotrichia,1.20%)、普氏菌(Prevotella,1.20%)、消化链球菌(Peptostreptococcus,1.20%)和颗粒链菌 (Granulicatella,1.20%)。本研究检测出牙周病组的优势菌依次为肠杆菌科未分类菌(Enterobacteriaceaeunclassified,47.41%)、假单胞菌(Pseudomonas,15.06%)、鲍曼不动杆菌(Acinetobacter,12.07%)、芽孢杆菌(Bacillus,7.04%)、气单胞菌未分类菌(Aeromonadaceaeunclassified,4.46%)、奈瑟氏菌(Neisseria,2.53%)、嗜血杆菌属(Haemophilus,2.32%)、梭杆菌(Fusobacterium,2.25%)、葡萄球菌(Staphylococcus,2.15%)、乳酸乳球菌(Lactococcus,1.95%),比较分析表明青海汉族人群中,牙周病组可疑的致病微生物类群及丰度比例与已有诸多报道不一致,优势属群亦明显不同。其原因可能有以下几点:①由于青海地区环境、气候特征和饮食习惯的明显差异造成的,青海地处高海拔地区,此环境下机体各系统各器官都产生不同程度的缺氧,使牙周厌氧菌生长繁殖加快。在低氧环境下,口腔内唾液分泌减少,口腔自洁功能减弱、加速了厌氧细菌的生长和菌群的形成;②可能与饮食有关,以面食为主,牛羊肉类和奶制品丰富;③青海省一直以来就是我国经济发展较为落后的地区之一,对口腔保健方面认识欠缺,因此,有必要进一步加强口腔卫生健康教育、增强居民口腔保健意识;④本研究取样仅选择唾液样本,而优势菌在唾液样本中量少而未检出或检出率低,后续研究将进一步改进。
综上述,本研究通过高通量测序技术初步对当地汉族牙周病人群优势菌的认识,可见牙周位点占据优势地位的是革兰阳性菌而非革兰阴性菌,亦初步揭示了此人群口腔疾病相关的微生物特征,其相关微生物菌群发生的显著变化较多,而正常口腔内的微生物菌群变化相对保守。
