以横纹肌溶解为表现的脂质沉积性肌病临床、病理与基因改变特点
2016-11-10漆学良丁卫江
姜 涛, 熊 李, 漆学良, 丁卫江
以横纹肌溶解为表现的脂质沉积性肌病临床、病理与基因改变特点
姜 涛, 熊 李, 漆学良, 丁卫江
目的 研究以横纹肌溶解(rhabdomyolysis,RM)为表现的脂质沉积性肌病(lipid storage myopathies,LSM)临床、病理及基因改变特点,提高对其诊断的警惕性。方法 回顾分析我院诊断的3例临床表现为急性横纹肌溶解的脂质沉积性肌病临床、病理及基因改变特点,结合相关文献复习进行讨论。 结果 2例以急性横纹肌溶解为首发表现,1例是诊断为LSM后,因停药后出现上呼吸道感染诱发急性横纹肌溶解。3例患者肌肉病理HE染色均可见筛状空泡,ORO染色证实肌纤维内有大量脂滴。其中2例可见较严重的肌纤维坏死。其中2例患者行ETFA、ETFB、ETFDH基因突变检测,ETFA基因正常,均发现ETFB基因第5外显子c. 734C>T(Thr245Met)和ETFDH基因第2外显子c. 92 C>T(Thr3Ile)杂合突变。结论 不明原因及反复出现肌肉酸痛、无力伴肌酸激酶明显增高、肌红蛋白尿等横纹肌溶解表现时,应考虑LSM可能,及时行肌肉活检明确诊断。以RM为表现的LSM出现相关ETFB与ETFDH基因的杂合突变,可能为多态性位点,意义未知。LSM患者停药或合并上呼吸道感染可诱发急性横纹肌溶解。
脂质沉积性肌病; 横纹肌溶解; 病理; 基因
脂质沉积性肌病(lipid storage myopathies,LSM)是由于影响脂质代谢的酶或肉毒碱缺乏,直接或间接干扰肌纤维内脂质代谢而导致肌纤维细胞内脂肪沉积的一组代谢性肌病,临床表现以肌无力和运动不耐受多见[1]。横纹肌溶解(rhabdomyolysis,RM)是由于肌纤维细胞膜破坏,肌纤维溶解,肌细胞成分进入血液和尿液[2]。近年来国内确诊的LSM病例数逐年增加,但表现为RM的LSM患者目前报道罕见,结合我院确诊的3例患者,对其临床、病理及基因突变检测资料分析如下。
1 资料
1.1 患者,女性,40岁,汉族。患者于2015年3月20日因“突发肌肉酸痛2 d”入院。患者2 d前无诱因出现背部及颈部肌肉酸痛,1 d后症状加重,出现全身肌肉酸痛,活动后加重,并出现棕色尿,但肢体活动尚正常。为进一步诊治收入院。患者发病2 m前曾反复出现四肢肌肉酸痛,易疲劳,因症状轻微,未引起重视。患者既往曾有“肾炎”病史,否认毒物、化学物质接触史,无劳累、不洁饮食、口服他汀类药物史。体格检查:四肢肌力Ⅴ-级,肌张力适中,全身肌肉酸痛,压痛明显,双侧腱反射稍减弱,双侧病理征阴性,脑膜刺激征阴性。入院后完善相关检查,甲状腺功能、肾功能、电解质基本正常,肝功能ALT 249.2 U/L,AST 853.9 U/L,心肌酶谱:CK 39949.2 U/L,CK-MB 577.91 U/L,LDH 1201.0 U/L,肌红蛋白3479.54 ng/ml。ANA、ENA、ANCA谱均阴性。入院后出现胸闷、气促,胸部CT示双肺感染,双肺下叶实变病灶为主,双侧胸腔积液,复查心肌酶谱:CK 70564.9 U/L,CK-MB1270.31 U/L,LDH3774.1 U/L,肌红蛋白5751.79 ng/ml,血气分析提示:pH 7.21,PO2148 mmHg,PaCO293 mmHg,BE 5.7 mol/L。治疗予补液、抗感染治疗,无创呼吸机辅助呼吸,行床旁连续肾脏替代疗法(continuous renal replacement therapy,CRRT)清除肌酶、毒素及炎症介质,同时丙种球蛋白(25 g/d×3 d)联合甲强龙冲击治疗(500 mg/d×4 d)。患者胸闷、肌肉酸痛较前好转,1 w后患者感肌肉酸痛明显改善,复查肌酶谱:CK 889.2 U/L,LDH 613.0 U/L,CK-MB 36.11 U/L,肌红蛋白228.99 ng/ml。2015年4月4日行左肱二头肌活检:HE染色示肌纤维大小基本一致,部分肌纤维可见大小不一的筛状空泡,可见部分坏死肌纤维,个别肌纤维可见核内移,未见灶性炎性细胞浸润,小血管结构未见异常(见图1)。ORO染色肌纤维内见大量脂滴聚集,少数融合成片(见图2)。改良Gomri(MGT)染色未见典型不整红边纤维;过碘酸-希夫(PAS) 染色见空泡纤维糖原轻度增多;还原型辅酶Ⅰ四氮还原酶 (NADH-TR)染色示空泡纤维以Ⅰ型为主。根据患者的临床表现、肌电图和肌肉病理检查结果,确诊为LMS。治疗予维生素B210片/次,3次/d。22 d后出院(4.11)复查肌酶谱:CK 166.9 U/L,LDH332.9 U/L,CK-MB15.20 U/L,肌红蛋白84.58 ng/ml。出院时患者四肢肌力正常,肌肉酸痛感消失。1 m后随访,患者肌力正常,无肌肉酸痛,无肌疲劳。
1.2 患者,男性,30岁,汉族。患者于2010年3月8日因“上腹痛胀、反复呕吐咖啡色液体半天”入南昌市第二医院普外科住院,诊断 “上消化道出血”,经禁食、抑制胃酸分泌及对症治疗后症状缓解,但1 w后出现双下肢肌痛和无力,进行性加重,蹲起、上楼及行走困难,并渐波及双上肢及颈部,双上肢不能上举,持物不稳,抬头困难,咀嚼及吞咽困难,并出现一过性棕色尿。患者既往有 “十二指肠球部溃疡”,多次因上消化道出血住院治疗。患者2 y前开始出现运动不能耐受,持续做家务30 min左右即感疲劳,曾诊断“抑郁症”,给予抗抑郁药(具体不详)治疗后无明显疗效。无家族史。入院查体:双侧眼轮匝肌、咬肌、颈肌肌力Ⅳ级,咳嗽声弱,进食有时呛咳,悬雍垂居中,两侧软腭运动可,四肢近端肌力Ⅱ级,远端Ⅳ级;四肢肌肉轻-中度肌萎缩,肌张力下降,腱反射减弱,病理反射未引出。实验室检查:肝肾功能、尿常规大致正常。肌酶谱:CK 14927 U/L,LDH 4002 U/L,CK-MB 225 U/L。肌电图示:肌源性损害。3月23日行右肱二头肌活检,HE染色示肌纤维大小基本一致,可见大量筛状空泡肌纤维及坏死肌纤维,未见肌分裂、核内移及肌核增多,肌内衣轻度增加,未见灶性炎性细胞浸润,小血管结构未见异常(见图3)。ORO染色肌纤维内见大量脂滴(见图4)。改良Gomri(MGT)染色未见典型不整红边纤维;过碘酸-希夫(PAS) 染色见空泡纤维糖原轻度增多;还原型辅酶Ⅰ四氮还原酶 (NADH-TR)染色示空泡纤维以Ⅰ型为主。根据患者的临床表现、肌电图和肌肉病理检查结果,确诊为LMS。予糖皮质激素、左旋肉碱治疗及维生素B2口服。治疗1 w后患者肌痛及肌无力症状迅速改善,2 w后复查肌酶谱:CK 79 U/L,LDH 602 U/L,CK-MB 22 U/L。随访至6 m,患者基本恢复,四肢近端肌力Ⅴ-级,远端肌力Ⅴ级。
1.3 患者,女性,34岁,汉族。患者2009年因“颈部及四肢近端乏力、疼痛”就诊我院,行肌肉活检HE染色示肌纤维大小基本一致,可见大量筛状空泡肌纤维,未见肌分裂、核内移及肌核增多,肌内衣轻度增加,未见灶性炎性细胞浸润,小血管结构未见异常。ORO染色肌纤维内见大量脂滴,提示脂质沉积性肌病。行ETFA、ETFB、ETFDH基因突变检测,ETFA基因正常,发现ETFB基因第5外显子c. 734C>T(Thr245Met)和ETFDH基因第2外显子c. 92 C>T(Thr3Ile)杂合突变(见图5、图6),治疗上予维生素B2、辅酶Q10口服,1 m后复查四肢肌力基本正常,肌酶谱:CK 50 U/L,LDH 201 U/L,CK-MB 25 U/L。继续服用维生素B2、辅酶Q10,半年后患者自行停服,2014年1月24日患者着凉后出现咽痛,伴发热,体温达37.8 ℃,2 d后出现四肢无力,以双下肢明显,逐渐加重至双下肢行走困难,双上肢持物不能,伴棕色尿。入院查体:双上肢肌力Ⅳ级,双下肢近端肌力Ⅰ级,远端Ⅱ级;肌张力稍减低。双下肢腱反射偏低,双侧病理征阴性,脑膜刺激征阴性。实验室检查:生化示肝、肾功能、电解质基本正常。心肌酶谱:CK 15849 U/L,LDH 5980.5 U/L,CK-MB 292.46 U/L,肌红蛋白3251.79 ng/ml。入院后考虑患者脂质沉积性肌病,因停药半年伴着凉感冒后出现急性横纹肌溶解,导致症状突发加重,予左卡尼汀、维生素B2、辅酶Q10等治疗,10 d后患者肌无力症状明显改善,双上肢肌力恢复至Ⅴ-原级,双下肢肌力Ⅳ级。出院后继续服用维生素B2,1 y随访,患者未再发作肌无力、肌疲劳。
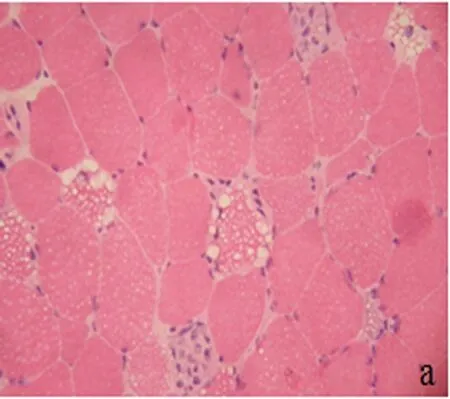
图1 部分肌纤维可见筛状空泡纤维并肌纤维坏死(HE染色×200)
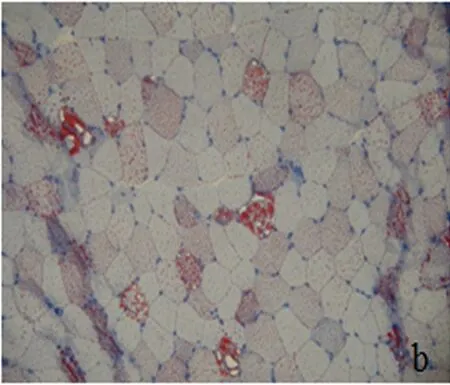
图2 大量肌纤维内脂滴显著增多(ORO染色×200)
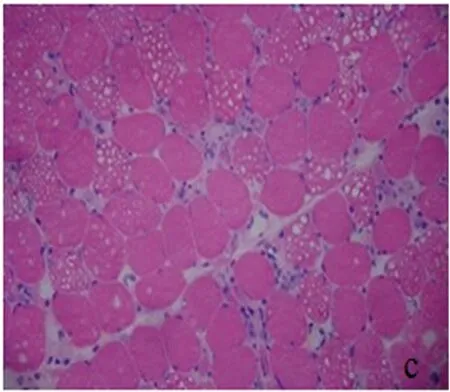
图3 大量筛状空泡纤维并肌纤维坏死(HE染色×200)
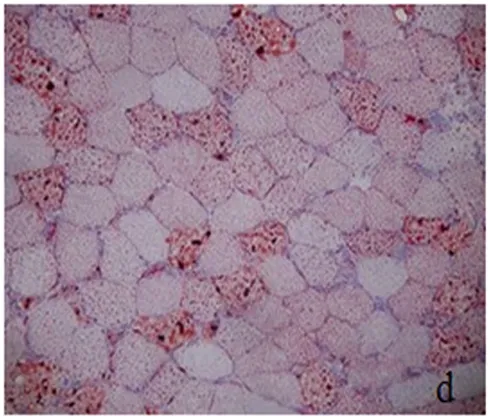
图4 大量肌纤维内脂滴显著增多(ORO染色×200)
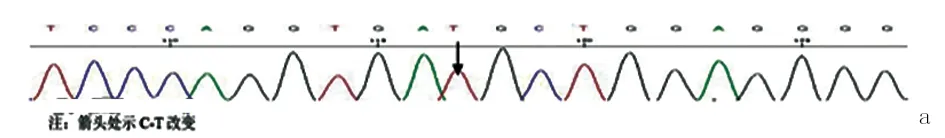
图5 ETFB基因第5外显子c. 734C>T(Thr245Met) 突变
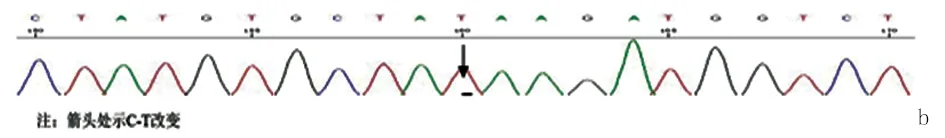
图6 ETFDH基因第2外显子c. 92 C>T(Thr3Ile)突变
2 讨 论
脂质沉积性肌病(lipid storage myopathies,LSM)是由于脂肪代谢障碍致异常增多的脂滴堆积在肌纤维内而引起的一组病理综合征。自从1973年首次描述以来,已发现在线粒体脂质代谢中18种酶及转运蛋白功能异常与脂质代谢障碍疾病相关。包括原发性肉毒碱缺乏症(primary carnitine deficiency,PCD)、肉毒碱棕榈酰转移酶缺乏症(carnitine palmitoyltransferase deficiency,CPTD)、极长链酰基辅酶A脱氢酶缺乏症(very long chain acyl-CoA dehydrogense deficiency,VLCADD)、线粒体三功能蛋白缺乏症(mitochondrial trifunctional protein deficiency,MTPD)、中链酰基辅酶脱氢酶缺乏症(medium-Chain Acyl-CoA dehydrogenase deficiency,MCADD)、多酰基辅酶脱氢酶缺乏症(multiple Acyl-CoA dehydrogenase deficiency,MADD)、肌肉辅酶Q10缺乏症及中性脂质沉积病(Neutral lipid storage diseases,NISDs)[3]。LSM典型的临床表现是发作性近端肌无力,易疲劳和运动不耐受。但有些LSM亚型则以反复横纹肌溶解为主要临床特点,脂质代谢障碍疾病以急性横纹肌溶解为主要特征的有β氧化障碍、CPTD、VLCADD[1]。其中肉毒碱棕榈酰转移酶 Ⅱ缺乏症是LMS最常见亚型,表现为反复发作的肌痛、肌无力、肌红蛋白尿,严重者可伴横纹肌溶解,多由运动、感染、禁食诱发[4]。 其余类型表现为急性横纹肌溶解者少见。
我们报道的3例病例中,第1例是以全身肌肉酸痛、肌红蛋白尿等横纹肌溶解为首发表现就诊,第2例是在上消化道出血后长时间禁食出现四肢无力、酸痛伴肌红蛋白尿等横纹肌溶解表现,这2例患者虽然均以横纹肌溶解为首发表现,但均在发病前有活动后轻微易肌疲劳、肌肉酸痛等代谢性肌病表现,排除其他易发生急性横纹肌溶解的疾病后,经肌肉活检病例证实为LSM。最后1例是在病理确诊为脂质沉积性肌病2 y,停药后因上呼吸道感染诱发横纹肌溶解。
LSM的治疗主要包括左卡尼汀、B族维生素和中小剂量皮质类固醇激素。国内外文献均报道皮质类周醇激素对LSM有肯定疗效,但作用机制不明。临床上核黄素治疗不仅与泼尼松具有同等疗效,而且可避免使用泼尼松可能带来的副作用,因此核黄素可作为目前治疗LSM的首选药物。我们3例患者1例经口服维生素B2,其余2例患者予维生素B2、左卡尼汀等治疗后症状都明显改善。
我们的3例患者均成年起病,2例以肌酸激酶明显增高、肌红蛋白尿等横纹肌溶解特点为临床首发表现,病理活检HE染色见大量筛状空泡肌纤维及坏死肌纤维,ORO染色肌纤维内见大量脂滴,维生素B2治疗有效,考虑诊断为临床表现为横纹肌溶解的LSM,可能为核黄素反应性MADD(riboflavin-responsive MADD,RR-MASDD),需要进一步研究,这与既往报道RR-MASDD可表现为横纹肌溶解症发现类似[5]。我们2例患者发现ETFB、ETFDH基因杂合突变,根据SNP 数据库报道,ETFDH 外显子2,Thr31Ile 与ETFB基因外显子5,Thr245Met为多态位点,可惜没找到致病突变,以后可借助二代测序等技术进一步查找致病基因。
过去由于对脂质沉积性肌病的认识不足,往往认为是LSM合并横纹肌溶解[6],现在我们认识到有部分亚型LSM即可以横纹肌溶解为主要表现,且多由运动、感染、禁食诱发。LSM出现横纹肌溶解的机制可能是由于脂肪酸氧化障碍引起ATP的耗竭。正常情况下,肌肉处休息状态时维持低水平钙浓度,肌纤维内钙浓度是由许多通道和泵调控,当ATP耗竭时通道和泵出现功能障碍,使肌细胞钙离子浓度持续升高,进而出现肌肉持续收缩和能量耗竭以及激活钙依赖性蛋白酶类和磷脂酶,结果导致肌原纤维、 细胞骨架和膜蛋白被破坏,最终肌细胞崩解[7]。
LSM肌电图表现并非如肌炎及其他类型肌病以肌源性损害多见,而多数表现为正常,甚至神经源性损害[8]。LSM合并周围神经受损也比较常见,周围神经受累可使肌电图失去特征性肌源性损害的特点,甚至出现神经源性损害的征象。周围神经受损的机制可能与脂质累及雪旺细胞有关[9]。我们报道的3例LSM患者有1例行肌电图检查,表现为肌源性损害。这提示与横纹肌溶解为临床表现的LSM,因肌肉严重受损,肌电图往往会出现或合并出现肌源性损害。
3例患者肌肉活检病理提示大量筛状空泡肌纤维及坏死肌纤维,ORO染色肌纤维内见大量脂滴。一般认为LSM患者病理仅见肌纤维内大量脂肪堆积,LSM结合临床和病理特点可分为2组,第1组为急性或亚急性起病,四肢近端肌无力,肌酶谱明显升高,对激素治疗敏感;第2组呈慢性迁延性病程,表现肌无力和对运动不耐受,对激素治疗不敏感,肌纤维内线粒体异常比较明显[10]。第1组类似多发性肌炎患者可能为潜在易出现横纹肌溶解的LSM,肌肉病理可见空泡肌纤维内脂滴明显增多,部分合并较严重肌纤维坏死,部分血清肌酶明显升高LSM患者病理上并未见到明显肌纤维坏死[11,12]。我们发现以急性横纹肌溶解为表现的LSM往往伴随坏死肌纤维,如果临床血清肌酶升高,但未表现横纹肌溶解,但病理出现较多坏死肌纤维的LSM患者,提示可能为亚临床横纹肌溶解,有急性横纹肌溶解的潜在风险。
临床上当患者表现为肌肉无力酸痛,肌红蛋白尿等横纹肌溶解表现时,在排除急性创伤以及常见非创伤性病因如缺血、代谢紊乱、感染、食物(小龙虾等)、药物如他汀类药物、乙肝抗病毒药物等中毒所导致[2],特别是当患者有活动后轻微易肌疲劳、肌肉酸痛等代谢性肌病表现时,应当考虑LSM可能,应及时行肌肉活检明确诊断,以免误诊导致患者不能得到及时正确的治疗。患者诊断LSM后,在运动、感染、禁食等诱因易出现急性横纹肌溶解,临床上需要引起重视。
[1]Liang WC,Nishino I. Lipid storage myopathy[J]. Curr Neurol Neurosci Rep,2011,11(1):97-103.
[2]Bosch X,Poch E,Grau J. Rhabdomyolysis and acute kidney injury[J]. N Engl J Med,2009,361( 1):62-72.
[3]李学明,丁卫江. 脂质代谢性疾病的临床和分子遗传学进展[J]. 中风与神经疾病杂志,2015,32(3):286-288.
[4]Bonnefont JP,Demaugre F,Prip-Buus C,et al. Carnitine palmitoyltransferase deficiencies[J]. Mol Genet Metab,1999,68:424-440.
[5]Izumi R,Suzuki N,Nagata M,et al. A case of late onset riboflavin-responsive multiple acyl-coa dehydrogenase deficiency manifesting as recurrent rhabdomyolysis and acute renal failure[J]. Intern Med,2011,50:2663-2668.
[6]赖 鸿,陈 恬,丁卫江. 脂质沉积性肌病合并横纹肌溶解的临床与病理特征[J]. 中国神经精神疾病杂志,2011,28(7):421-422.
[7]Warren JD,Blumbergs PC,Thompson PD. Rhabdomyolysis:a review[J]. Muscle & Nerve,2002,25(3):332-347.
[8]龚凌云,胡 凡,黄 刚,等. 脂质沉积性肌病临床与神经电生理分析[J]. 中国神经免疫学和神经病学杂志,2013,20(1):6-8.
[9]崔丽英,汤晓芙,袁 静,等. 脂质沉积性肌病合并周围神经病变的临床和神经电生理研究[J]. 中国神经免疫学和神经病学杂志,1998,5:157.
[10]陈 琳,郭 重,郭玉璞,等. 脂质沉积性肌病的临床和病理特点[J]. 中华神经科杂志,1998,31:165-167.
[11]陈 琳,郭玉璞,任海涛,等. 貌似多发性肌炎的脂质沉积性肌病病理改变[J]. 中华神经科杂志,2001,2:81-83.
[12]管玉青,谢作善,郑 卉,等. 误诊为多发性肌炎的脂质沉积性肌病-附3例报告[J]. 中国神经精神疾病杂志,2012,38:101-104.
Clinical,pathological and genetic features of lipid storage myopathy presenting as rhabdomyolysis
JIANGTao,XIONGLi,QIXueliang,etal.
(DepartmentofNeurology,TheSecondAffiliatedHospitaltoNanchangUniversity,Nanchang330006,China)
Objective Some cases of lipid storage myopathy (LSM) are present as rhabdomyolysis. To study the clinical,pathological and genetic features is helpful to diagnosis the LSM presenting as rhabdomyolysis. Methods Three cases of LSM presenting as acute rhabdomyolysis were analyzed on clinical,pathological and genetic features. The related literatures were reviewed. Results Two cases were present as rhabdomyolysis firstly. In one case who had been diagnosed as LSM,rhabdomyolysis was induced by the upper respiratory tract infection after drug withdrawal. Electrophysiology showed myogenic damage in one case,while the others were normal. All of the muscle specimens showed typical accumulation of lipid in muscle fibers. Necrotic fibers were found in 2 cases. Two patients performed ETFA、ETFB、ETFDH genetic testing. ETFA genetic testing was normal,while both the ETFB gene analysis identified a c. 734C>T (Thr245Met) heterozygous mutation and ETFDH gene analysis identified a c. 92C>T (Thr3Ile) heterozygous mutation. Conclusion When patients occur as muscle pain and fatigue accompany with high level of CK in serum and myoglobinuria repeatedly,LSM should be considered. Muscle biopsies and histochemical studies are necessary to diagnosis LSM. Clinical significance of two heterozygous mutation of the gene is unknown. Two heterozygous mutations may be polymorphic loci. Stop taking the drugs and have a cold may be the risk factors for LSM patients to have acute rhabdomyolysis.
Lipoidosis; Rhabdomyolysis; Pathology; Gene
1003-2754(2016)01-0060-04
2015-11-19;
2015-12-28
(南昌大学第二附属医院神经内科,江西 南昌 330000)
漆学良,E-mail:qixueliang766@163.com
R746
A
