他汀相关免疫介导性坏死性肌病2例临床病理及文献复习
2015-04-08文琼芳金京玉刘立芝蒲传强
文琼芳,金京玉,赵 征,朱 剑,刘立芝,班 瑞,蒲传强,黄 烽
(中国人民解放军总医院风湿科,北京 100853)
免疫介导性坏死性肌病(immune-mediated necrotising myopathy,IMNM)是一种较常见的自身免疫性肌病,发病率仅次于皮肌炎(dematomyositis,DM)、多肌炎(polymyositis,PM)[1],临床表现为肌痛、肌无力、肌痉挛等,可继发于结缔组织病、肿瘤等。目前该病发病机制尚不明确,文献报道其多与环境、病毒感染、药物等因素相关[1]。其中,报道最多的是他汀类药物,一些患者服用他汀类药物后可发生DM、PM或IMNM[2]。因受限于肌肉病理、免疫组化染色等实验室检查手段,目前国内尚无他汀诱发IMNM的报道,仅有少量IMNM病例报道[3-4]。现报道2例中国人民解放军总医院风湿科于2014年收治的经肌肉活检证实的服用他汀类药物后诱发的IMNM患者,并结合文献分析如下。
对象和方法
资料
回顾性分析2例2014年9月至11月在中国人民解放军总医院行肌肉活检,且病理诊断为坏死性肌病,并排除结缔组织病、肿瘤、内分泌系统疾病及其他炎症性肌病的患者。2例患者均进行了如下检查:血生化、红细胞沉降率、C反应蛋白、巨细胞病毒抗体、EB病毒抗体、抗核抗体、抗Jo-1抗体、抗线粒体抗体、抗Mi-2抗体、抗中性粒细胞胞浆抗体(anti-neutrophil cytoplasmic antibody,ANCA)、甲状腺功能7项、肿瘤标记物、PET-CT、肌电图、肌肉病理。
电生理学检查
应用Keypoint(丹麦产)肌电图仪进行同轴单心针电极肌电图检查。检查内容包括:静息状态下自发电位、轻收缩时运动单位电位、重收缩时运动单位电位、运动神经传导速度、感觉神经传导速度。
病理学检查
2例患者均签署知情同意书,并进行了肌肉活检(均为股四头肌)。肌肉组织冰冻切片除进行常规染色(HE、改良GomoriTrichrome染色、高碘酸Schiff反应、油红O与苏丹黑染色)和酶组织化学染色(还原型辅酶Ⅰ四氮唑还原酶、酸性磷酸酶、琥珀酸脱氢酶、三磷酸腺苷酶、非特异酯酶染色),以及CD4、CD8、CD68、C5B9、主要组织相容性复合体-Ⅰ染色。
结 果
临床特点
2例均为女性、亚急性起病,例1、2的发病年龄分别为55和63岁,病程分别为5和3个月。例1服用阿托伐他汀(10 mg/d)2个月后,因肌酶水平升高改为瑞舒伐他汀10 mg/d;服用4个月,因出现吞咽困难,于入院5个月前停用他汀类药物。例2服用辛伐他汀10 mg/d半年,因出现肌无力症状,于入院2个月前停用。2例患者均合并高胆固醇血症(表1)。
实验室检查
肌酸激酶:例1最高达3 126 U/L(参考范围2~200 U/L),例2最高达8 780 U/L。PET-CT检查:例1未见异常;例2检查提示躯干部多发淋巴结肿大伴轻度摄取(左腋下、腹膜后、肠系膜),淋巴结穿刺活检证实为反应性增生。
2例ESR、CRP、补体、免疫球蛋白、巨细胞病毒抗体、EB病毒抗体、抗核抗体、抗Jo-1抗体、抗线粒体抗体、抗Mi-2抗体、ANCA、甲状腺功能7项等均正常。肿瘤标记物:例1正常;例2 NSE升高,为30.4 ng/ml(参考值范围0~24.0 ng/ml)。
肌肉组织电生理检查
2例患者均为肌源性损害。
肌肉病理检查
位置均为有明显肌无力症状的股四头肌。
例1:可见一些肌纤维变性和坏死,但吞噬现象不明显,伴有少数核内移纤维,无肌纤维肥大,肌纤维间隙增宽。血管形态正常,未见镶边空泡纤维和炎症细胞(图1)。改良GomoriTrichrome染色:无破碎红纤维。还原型辅酶Ⅰ四氮唑还原酶染色:部分肌纤维结构破坏。琥珀酸脱氢酶染色:肌纤维琥珀酸脱氢酶活性正常。酸性磷酸酶染色:可见坏变肌纤维酸性磷酸酶活性增强。油红O染色:阴性。苏丹黑染色:阴性。高碘酸Schiff反应:阴性。神经元特异性烯醇化酶染色:阴性。三磷酸腺苷酶染色:肌纤维分型良好,两型均受累,无群组化现象。免疫组化结果未提示CD4+CD8+T淋巴细胞及CD68+巨噬细胞浸润,未见C5b-9、主要组织相容性复合体-Ⅰ染色。
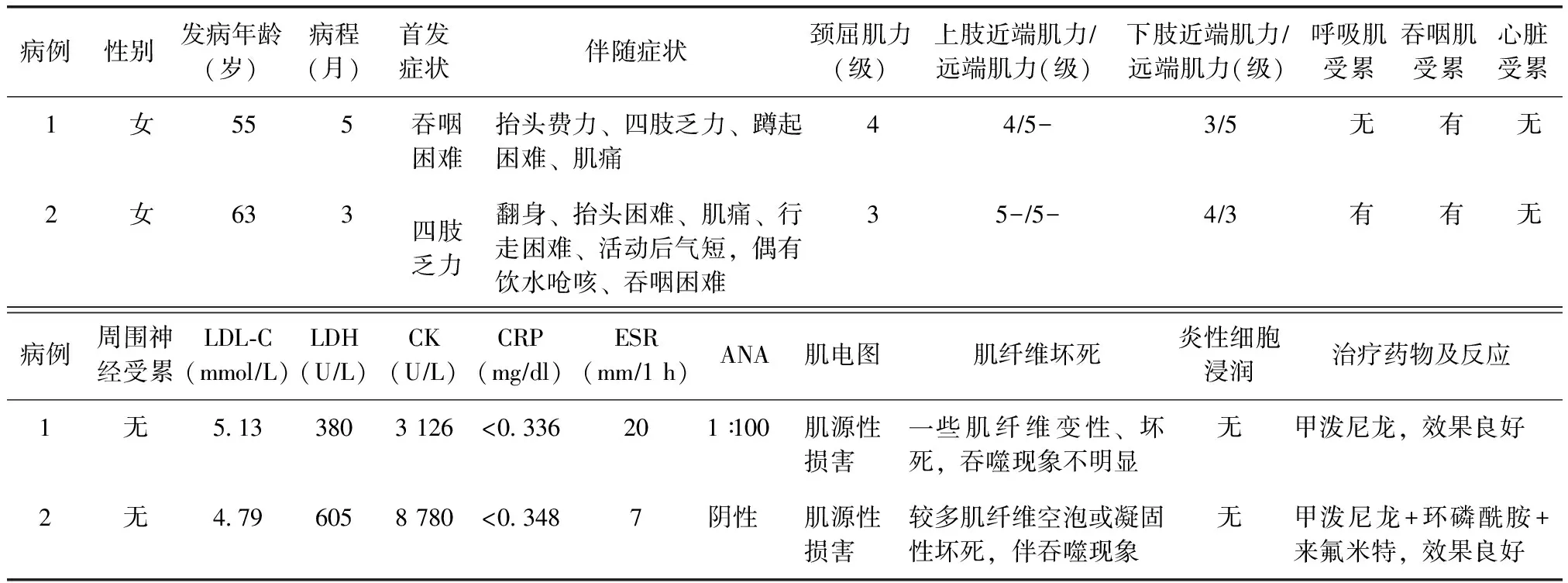
表1 2例他汀相关免疫介导性坏死性肌病患者的临床、病理特点
LDL-C:低密度脂蛋白胆固醇;LDH:乳酸脱氢酶;CK:肌酸激酶;CRP:C反应蛋白;ESR:红细胞沉降率;ANA:抗核抗体
例2:可见较多的肌纤维空泡和凝固性坏死,伴吞噬现象,无炎性细胞浸润,肌纤维肥大不明显,肌纤维间隙增宽,未见核内移肌纤维。血管形态正常,未见镶边空泡纤维(图2)。改良GomoriTrichrome染色:无破碎红纤维。还原型辅酶Ⅰ四氮唑还原酶染色:肌纤维结构破坏。琥珀酸脱氢酶染色:肌纤维琥珀酸脱氢酶活性正常。酸性磷酸酶染色:肌纤维及其间隔酸性磷酸酶活性增强。油红O染色:阴性。苏丹黑染色:阴性。高碘酸Schiff反应:阴性。神经元特异性烯醇化酶染色:较多坏变肌纤维阳性。三磷酸腺苷酶染色:肌纤维分型良好,无群组化现象。
诊断及鉴别诊断
2例患者临床表现及实验室检查结果相似,主要表现为四肢肌力下降,伴肌酶明显升高,无皮肤损害。在鉴别诊断时,充分考虑了特发性炎症性肌病、肌营养不良症、代谢性肌病、甲状腺功能减退性肌病、中毒性肌病、类固醇肌病、肿瘤性疾病等。结合这2例患者的病例特点,重点考虑炎症性肌病的鉴别。PM起病隐袭,常有对称性近端肌无力、肌压痛,偶有吞咽困难、发音不清,呼吸肌受累可出现呼吸困难,少数患者合并肺间质纤维化、心肌炎等。DM除有PM表现外,常有典型的Gottron征、披肩征、V型红斑及技工手等皮疹。IMNM也主要表现为四肢肌无力、肌痛,肌酶升高,病理诊断结果具有鉴别意义。IMNM以肌纤维坏死为主要病理特征,坏死形态多样,可表现为空泡、凝固、溶解、破碎以及玻璃样坏死等[1,3-4]。本组患者肌肉病理主要表现为肌纤维坏死,血管形态正常,无束周萎缩的特点。例2可见吞噬现象。例1免疫组化结果未见C5b9、主要组织相容性复合体-Ⅰ染色。2例患者既往均体健,服用他汀类药物后出现肌无力、肌痛等症状,停药后持续加重,结合这2例患者的临床表现、电生理和肌肉病理特点,认为均符合他汀相关IMNM的诊断。
治疗及随访情况
例1:甲泼尼龙30 mg/d治疗1周后症状明显好转,后逐渐减量至8 mg/d,3个月后复查四肢肌力恢复至5级,颈肌肌力5-级,肌酸激酶降至231.3 U/L。
例2:因症状较重,确诊后予醋酸泼尼松(45 mg/d,逐渐减量至15 mg/d)、环磷酰胺(每2周0.6 g,共2次)治疗。后因患者出现头晕、恶心、血压升高,将免疫抑制剂改为来氟米特10 mg/d。3个月后下肢肌力恢复至5级,颈肌肌力4级,肌酸激酶降至38.2 U/L,神经元特异性烯醇化酶降至8.95 ng/ml。
讨 论
IMNM是一种呈亚急性或隐匿性起病的肌病,最早报道于1969年[3]。1975年Bohan等首次详细描述了炎性肌病中皮肌炎和多肌炎的分类特征[1]。此后他们又发现,有些肌病患者肌肉活检特征表现为有大量肌纤维坏死,但缺乏炎性细胞浸润和吞噬细胞。2003年,欧洲神经肌肉研究中心将IMNM与皮肌炎、多肌炎等一起归类于特发性炎症性肌病(idiopathic inflammatory myopathy,IIM)[5]。现有研究已证实,IMNM是一种临床表现、病理基础均明显不同于皮肌炎、多肌炎,激素及免疫抑制剂疗效较好的肌病。进一步的研究将IMNM分为许多不同亚型,包括肿瘤相关,结缔组织病相关或病毒感染相关,还有与如抗合成酶抗体、抗信号识别颗粒(signal recognition particle,SRP)抗体、抗3-羟基-3-甲基戊二酰辅酶A还原酶(hydroxy-3-methyl-glutaryl-coa reductase,HMGCR)抗体等特异性自身抗体相关等亚型[1]。
他汀类药物是一种常用的心血管疾病治疗药物,具有调脂、抗炎、稳定动脉粥样斑块等作用,其药理学作用靶点为HMGCR。研究已经证实,他汀类药物及其他HMGCR抑制剂可以导致一系列的骨骼肌不良反应,既可以表现为轻度肌痛、肌无力等肌肉症状,伴或不伴肌酶增高,也可以表现为持续肌肉症状伴肌酶明显增高的炎症性肌病或坏死性肌病,甚至危及生命的横纹肌溶解[2,6]。一般而言,他汀诱发的骨骼肌不良反应较轻,通过停药、对症处理后可以迅速恢复,但出现停药后持续进展的情况时则需考虑IMNM可能。
1986年Reeves等[7]首次发现第一种IMNM相关抗体-抗SRP抗体。此后研究证实,该抗体阳性的患者具有一组完全不同的特征,如严重迅速进展的乏力,高度升高的肌酶水平,极少肌肉外表现,病理检查炎症细胞少见[8-9]。Christopher-Stine等[10]发现,在26例具有类似特征的既往未发现自身抗体的患者中,16例体内有一种能识别相对分子质量为200 000和100 000长度蛋白质的自身抗体——抗HMGCR抗体。抗HMGCR抗体虽然经常与他汀诱发的IMNM相关,但大多数服用他汀的患者体内没有抗HMGCR抗体,包括那些出现他汀肌毒性但停药后未自发进展者[11]。Mammen等[12]在750例自身免疫性肌病患者中发现,其中45例抗HMGCR抗体阳性,其中仅30例(67%)有他汀暴露史。也有研究认为,无他汀暴露史的抗HMGCR抗体阳性患者的临床表现明显不同,表现为更高的肌酶水平,白色人种少见[13]。遗憾的是,国内尚未开展抗HMGCR抗体的检测,本文报道的2例患者的抗HMGCR抗体无法检测。需注意的是,并不是所有的抗HMGCR抗体阳性和抗SRP抗体阳性的患者都具有排除性的肌肉病理结果。部分有他汀暴露史的抗HMGCR抗体阳性患者(11%)和无他汀暴露史的抗HMGCR抗体阳性患者(39%)外周血管周围有炎性细胞浸润[12]。国外报道,7例抗SRP抗体阳性患者中2例有肌膜内和(或)外周血管周围的淋巴细胞浸润[8],这与国内[4,9]的研究数据相符。
IMNM的发病机制尚未完全阐明,现有研究认为与他汀类药物、肿瘤、感染或免疫异常等有关[1,11,14]。同时,还发现了一些他汀相关性IMNM的易感基因[14],包括SLCO1B1(编码肝跨细胞膜他汀类转运蛋白有机阴离子运输多肽OATP1B1)、CYP2D6和CYP3A5(肝细胞色素酶P450 2D6和3A5是他汀代谢酶)、ABCB1(编码外流性转运糖蛋白P)、GTAM(编码参与肌酐合成的甘氨酸脒基转移酶)等。
治疗他汀相关IMNM时较治疗其他他汀类药物导致的肌肉损害须更为积极,大剂量激素、静注免疫球蛋白(intravenous immunogloblin,IVIG)治疗效果较好,许多患者需要联用免疫抑制剂治疗[3,4,9,12]。有他汀暴露史的抗HMGCR抗体阳性患者对IVIG治疗反应非常好,无他汀暴露史的抗HMGCR抗体阳性患者比有他汀暴露史的患者更难治疗[13]。难治性的抗SRP抗体阳性患者通常对利妥昔单抗治疗反应好,而无他汀暴露史的抗HMGCR抗体阳性患者则没有这种表现(个案报道)[15],这表明抗SRP抗体相关的IMNM与抗HMGCR抗体相关IMNM的似乎存在本质不同。但是这些回顾性分析研究和个案报道的结果有待进一步的试验来证实。研究发现大部分患者出现IMNM症状后会持续进展,甚至可能在撤药后首发[2],这与本组2例患者的表现相类似。因此,对于服用他汀类药物的患者需高度警惕肌痛、肌无力等不适,定期监测肌酶谱。目前文献报道,经激素和免疫抑制剂治疗后大多数患者能够缓解甚至痊愈[1,3,4,9],但因缺乏大样本的长期观察研究,该病的预后有待进一步研究。
从现有的研究来看,IMNM的疾病谱可能远远大于最初的认识,其包含了一系列由免疫系统异常导致的坏死性异质性肌肉病。不同专业领域专家,尤其是风湿病学家、神经病学家、肌病学家的合作将有助于总结分析他汀相关IMNM的流行病学、病理生理、病理特点等问题。
(本文图1、2见插页Ⅳ)
[1]Stenzel W,Goebel HH,Aronica E.Review:immune-mediated necrotizing myopathies-a heterogeneous group of diseases with specific myopathological features[J].Neuropathol Appl Neurobiol, 2012,38:632-646.
[2]Padala S,Thompson PD.Statins as a possible cause of inflammatory and necrotizing myopathies[J].Atherosclerosis,2012,222:15-21.
[3]刘芳,蒲传强,罗平,等.坏死性肌病9例临床与病理分析[J].卒中与神经疾病,2005,12:149-151.
[4]张英爽,孙阿萍,陈璐,等.免疫介导性坏死性肌肉病四例临床病理分析及随访[J].中华内科杂志,2015,54:35-39.
[5]Hoogendijk JE,Amato AA,Lecky BR,et al.119th ENMC international workshop:trial design in adult idiopathic inflammatory myopathies,with the exception of inclusion body myositis,10-12 October 2003,Naarden,The Netherlands[J].Neuromuscul Disord,2004,14:337-345.
[6]Jamal SM,Eisenberg MJ,Christopoulos S.Rhabdom-yolysis associated with hydroxylmethylglutaryl-coenzyme A reductase inhibitors[J].Am Heart J, 2004,147:956-965.
[7]Reeves WH,Nigam SK,Blobel G.Human autoantibodies reactive with the signal-recognition particle[J].Proc Natl Acad Sci USA,1986,83:9507-9011.
[8]Miller T,Al-Lozi MT,Lopate G,et al.Myopathy with antibodies to the signal recognition particle:clinical and pathological features[J].J Neurol Neuro-surg Psychiatry,2002,73:420-428.
[9]Wang L,Liu L,Hao H,et al.Myopathy with anti-signal recognition particle antibodies:clinical and histopathological features in Chinese patients[J].Neuromuscul Disord, 2014,24:335-341.
[10] Christopher-Stine L,Casciola-Rosen LA,Hong G,et al.A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy[J].Arthritis Rheum,2010,62:2757-2766.
[11] Mammen AL,Pak K,Williams EK,et al.Anti-HMG-CoA reductase antibodies are rare in statin users,including those with self-limited musculoskeletal side effects[J].Arthritis Care Res (Hoboken),2012,64:269-272.
[12] Mammen AL,Chung T,Christopher-Stine L,et al.Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy[J].Arthritis Rheum,2011,63:713-721.
[13] Werner JL,Christopher-Stine L,Ghazarian SR,et al.Antibody levels correlate with creatine kinase levels and strength in anti-HMG-CoA reductase-associated autoimmune myopathy[J].Arthritis Rheum,2012,64:4087-4093.
[14] Luo YB,Mastaglia FL.Dermatomyositis,polymyositis and immune-mediated necrotising myopathies[J].Biochim Biophys Acta, 2015,1852:622-632.
[15] Valiyil R,Casciola-Rosen L,Hong G,et al.Rituximab therapy for myopathy associated with antisignal recognition particle antibodies:a case series[J].Arthritis Care Res (Hoboken),2010,62:1328-1334.
