化妆品中挥发性有机溶剂的通用检测方法
2014-12-26黄湘鹭王钢力张庆生
达 晶, 黄湘鹭, 王钢力 , 曹 进, 张庆生
(中国食品药品检定研究院,北京100050)
化妆品中普遍存在有机溶剂残留,这些有机溶剂既可从原材料、生产过程中带入造成残留,亦可是化妆品生产者为改善产品的性能而人为地加入造成残留[1],如提取超氧化物歧化酶(SOD)时使用氯仿-乙醇溶剂,提取茶多酚时使用氯代甲烷(如氯仿、二氯甲烷)和乙酸乙酯[2]。普通指甲油的溶剂成分基本是有毒或者有害物质,其中对人体危害比较大的是邻苯二甲酸酯、甲醛、苯系物等。苯系物(苯、甲苯、乙苯、二甲苯、异丙苯、苯乙烯)是很多化工企业经常使用的有机溶剂,也是最常见的可引起人体中毒的一类物质[3]。《化妆品卫生规范》(2007)中规定:苯、氯仿、四氯化碳、二氯乙烷、二氯乙烯、四氯乙烯、乙腈等有机溶剂禁止用于化妆品生产;二氯甲烷为限用[4];而其他一些有毒的有机溶剂,如甲苯、二甲苯、乙苯等则没有标明禁用或限用。但是,即便是常用的有机溶剂,如异丙醇、乙酸乙酯、丙酮等,长期接触也会对人体产生毒害[5],因此有必要对化妆品中存在的有机溶剂种类和残留量予以评估和管理,并同时建立相应的检验检测技术用于质量管理。
由于化妆品生产中普遍使用有机溶剂,同时还存在违规使用的现象,因此成品中可能含有的溶剂种类繁多,现有针对具体目标分析物的检测方法往往不能满足实际检测需求。本方法以36 种常见或可能使用的溶剂为模板,从检测通用性的角度,采用气相色谱-质谱方法,建立溶剂残留筛查知识库,并进而建立定量方法用于化妆品中残留溶剂的筛查、鉴别和定量。该方法可用于化妆品中可能存在的多种溶剂残留种类鉴别和含量测定,通用型的测定和流程也可扩大至其他溶剂的筛查和测定。这一方面提高了化妆品中多溶剂残留测定在实际检测中的效率,另一方面也为全面建立化妆品中溶剂残留知识库,构建通用型筛查方法提供了途径。
1 实验部分
1.1 仪器与试剂
Agilent 7000 气相色谱-三重四极杆质谱仪,Agilent 7697A 顶空进样仪,分析天平,涡旋混合仪,顶空瓶(20 mL)。
乙醇、二氯甲烷、甲基叔丁基醚、乙酸乙酯、四氢呋喃、乙腈购自Fisher 公司;乙基苯、乙酸异丙酯、仲丁醇购自Fluka 公司;乙酸异丁酯、乙酸丁酯、对二甲苯、三氯乙烯、4-甲基-2-戊酮购自Sigma 公司;甲酸乙酯、乙酸甲酯、苯、1,2-二氯乙烷、2-丁酮、四氯乙烯、异丁醇、正丁醇、正戊醇、异戊醇、间二甲苯、乙酸异戊酯、1,4-二氧六环、乙酸丙酯、邻二甲苯购自国药集团化学试剂有限公司;乙醚、丙酮、氯仿、四氯化碳、异丙醇、正丙醇、甲苯购自北京化工厂;36 种标准品纯度均大于98.0%。甲醇、正戊烷、正己烷、正庚烷为色谱纯,购自Fisher 公司。C8 ~C40 正构烷烃混合标准溶液(溶剂为氯仿)购自AccuStandard公司。氯化钠(NaCl)为优级纯,购自国药集团化学试剂有限公司,使用前在马弗炉内550 ℃烘烤过夜。
1.2 标准溶液的配制
挥发性有机溶剂标准储备液:称取挥发性有机溶剂对照品约20 mg(精确至0.1 mg)于10 mL 容量瓶中(容量瓶中预先加入4 mL 甲醇,称量过程中不时轻摇容量瓶),立即以甲醇稀释并定容至10 mL,配成约2 g/L 的单标准溶液。储备液保存于-20℃冰箱中。吸取以上标准储备液适量,用纯水稀释至所需浓度的混合标准使用液和系列线性溶液。
正构烷烃混合标准溶液:分别吸取适量的正戊烷、正己烷、正庚烷及C8 ~C40 正构烷烃混合标准溶液,以甲醇稀释,得到500 mg/L 的正构烷烃混合标准储备液。吸取适量正构烷烃混合标准储备液以水稀释得到各组分质量浓度为0.1 mg/L 的正构烷烃混合标准溶液。
1.3 仪器工作条件
1.3.1 顶空条件
平衡温度为60 ℃;进样温度为100 ℃;传输线温度为120 ℃;平衡时间为30 min;定量环为1 mL。
1.3.2 色谱条件
极性柱为VF-1301ms 毛细管色谱柱(30 m ×0.25 mm×1 μm),非极性柱为DB-5ms 毛细管色谱柱(30 m ×0.25 mm ×1 μm);载气为氦气,纯度≥99.999%,流速为1.3 mL/min;进样口温度为150℃;进样方式为分流进样,分流比为50 ∶1;程序升温:初始温度30 ℃,保持10 min,以5 ℃/min 升至100 ℃,再以30 ℃/min 升至220 ℃,保持5 min。
1.3.3 质谱条件
电子轰击源,碰撞能量70 eV;离子源温度230℃;四极杆温度150 ℃;传输线温度220 ℃;选择离子监测(SIM)模式。36 种挥发性有机溶剂在极性柱和非极性柱的保留时间、定性离子和定量离子见表1。采用分时段分别监测,极性柱和非极性柱的质谱参数分别见表2 和表3。
1.4 双柱保留指数知识库
取正构烷烃混合标准溶液进行GC-MS 分析,用于各组分保留指数(KI)值的计算,分别记录各正构烷烃的保留时间,采用线性升温公式计算各组分的KI 值。筛查参数如表4 所示。
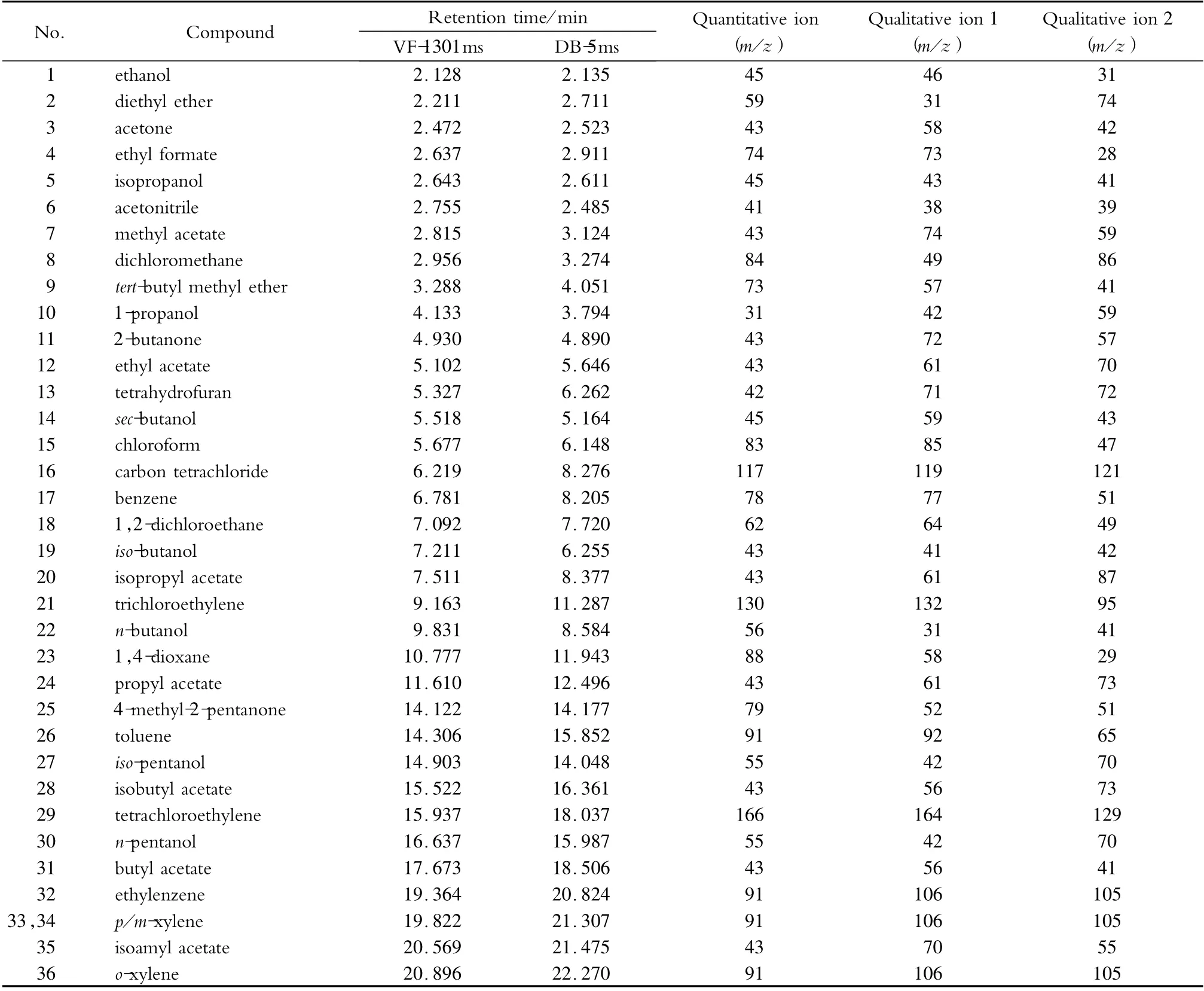
表1 36 种挥发性有机溶剂的GC-MS 检测参数Table 1 GC-MS parameters of the 36 volatile organic solvents
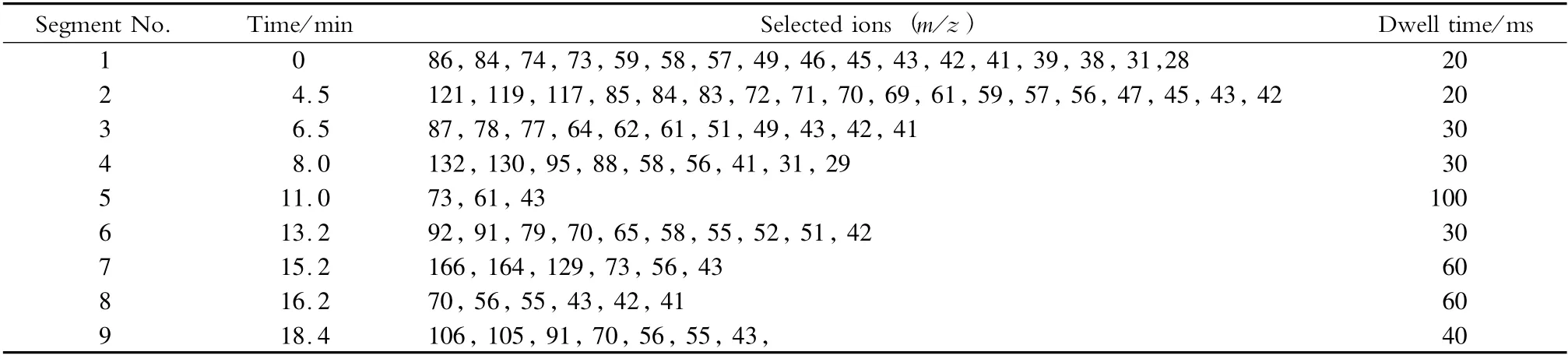
表2 36 种挥发性有机溶剂在选择离子监测模式下的质谱参数(极性柱)Table 2 Mass spectrometric parameters of the 36 volatile organic solvents under SIM mode (polar column)
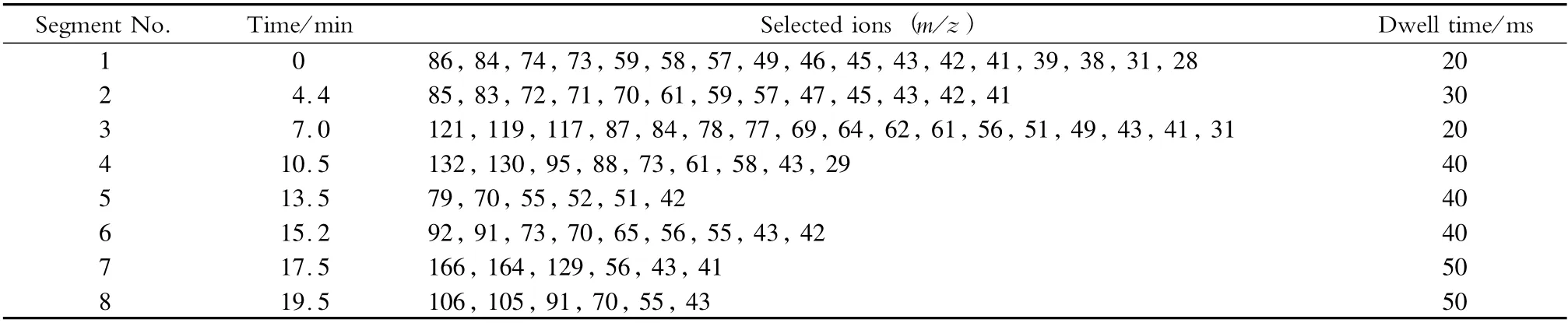
表3 36 种挥发性有机溶剂在选择离子监测模式下的质谱参数(非极性柱)Table 3 Mass spectrometric parameters of the 36 volatile organic solvents under SIM mode (nonpolar column)
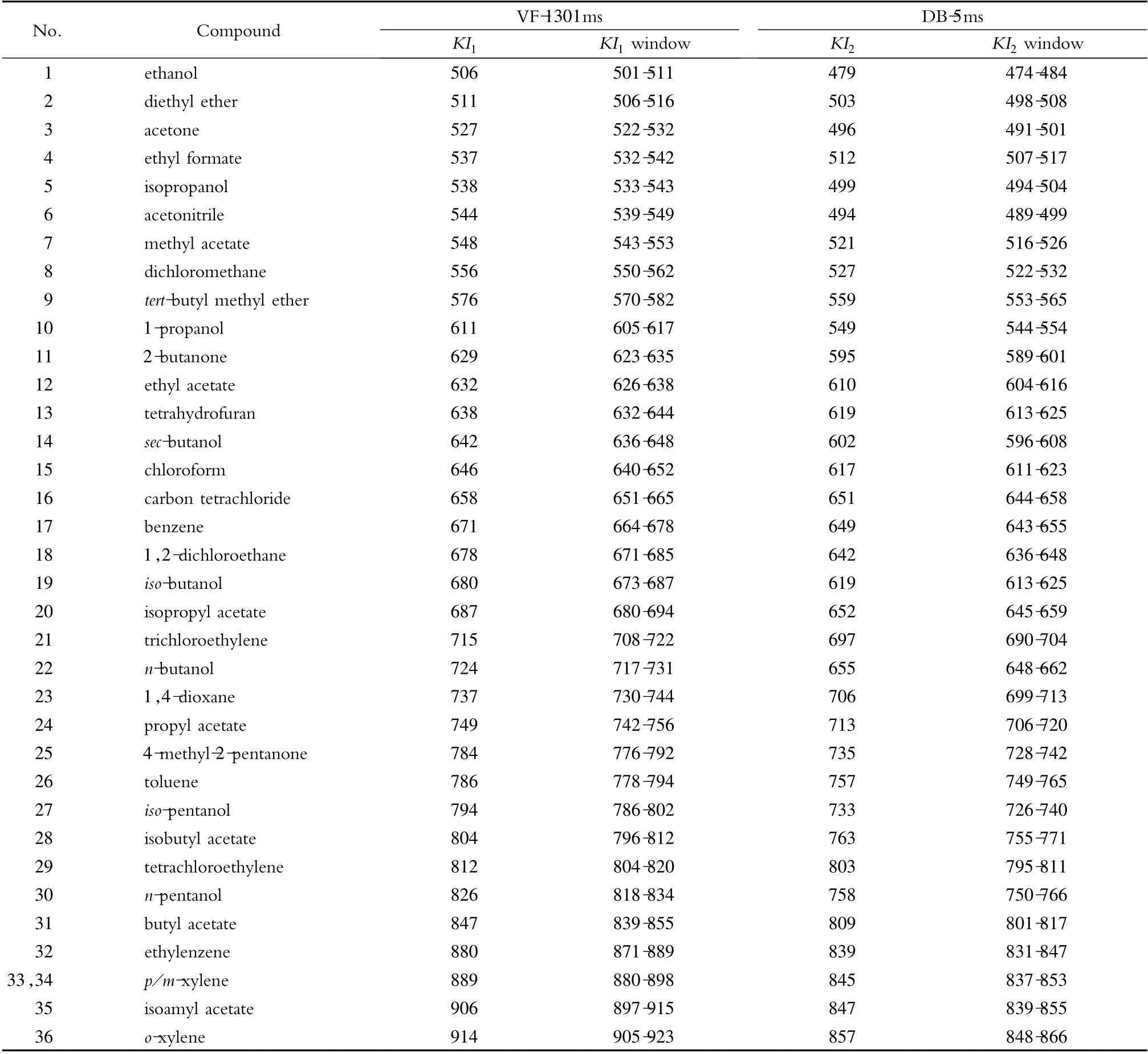
表4 36 种挥发性有机溶剂的保留指数Table 4 KI values of the 36 volatile organic solvents
1.5 样品测定
1.5.1 样品的初筛
称取样品约1.000 g(精确至0.001 g)于100 mL 容量瓶中,加水溶解并定容至刻度,对难溶于水的样品,加入适量甲醇助溶后用水定容至刻度(甲醇体积不超过定容体积的1%)。涡旋1 min。取10 mL 样品溶液至20 mL 顶空瓶中,加入1 g NaCl,加盖密封。
将样品注入气相色谱-质谱仪,分别测定样品在极性柱和非极性柱上的保留指数,与本项目自建的双柱保留指数知识库进行对比,若样品待测组分均在某溶剂双柱保留指数知识库数据范围内,则使用NIST 质谱库检索进一步筛查待测组分。若结果呈阳性,则使用相应溶剂对照品进行确证和定量计算。
1.5.2 结果确证及定量测定
1.5.2.1 初筛结果的确证和工作曲线绘制
分别吸取样品中疑似存在的挥发性有机溶剂标准储备液适量,用纯水稀释至所需浓度的混合标准使用液和系列线性溶液。选择VF-1301ms 毛细管色谱柱,按照指定条件同时测定对照品溶液和样品溶液,比较对照品溶液与样品溶液中色谱峰的KI值、离子丰度比,进一步确证初筛结果。同时根据系列线性溶液质量浓度和峰面积绘制工作曲线。
1.5.2.2 样品定量测定
按1.5.1 节方法配制样品溶液,选择VF-1301ms 毛细管色谱柱,在设定条件下测定样品溶液,根据工作曲线求出样品中所含挥发性有机溶剂的含量。
2 结果与讨论
2.1 研究对象及稀释溶剂的选择
根据文献调研和理化常数比较,选择在常见配方产品中使用的3 个沸点水平(即40、80、120 ℃)的36 种常见代表性挥发性有机溶剂作为研究模板。
对于挥发性有机溶剂的稀释溶剂的选择,文献报道主要有甲醇、丙酮、正己烷、N,N-二甲基乙酰胺(DMA)等[6]。甲醇和DMA 能与水、醇、醚、酯、烷烃和芳香类化合物等有机溶剂任意混合,但DMA在测定过程中出峰时间对其他待测物有干扰,因此选择甲醇作为36 种挥发性有机溶剂的稀释剂。
2.2 色谱柱的选择
由于不同物质在一个色谱系统中可能具有相同的保留值,因此定性结果往往不够准确,特别是对于未知化合物的定性。如果采用两个不同极性的色谱系统,则可大大提高定性分析结果的准确性。采用双柱定性时,所选的两根色谱柱的极性应具有较大差别,极性差别越大,定性分析结果可信度越高[7]。
本文选择了两根极性不同的毛细管色谱柱VF-1301ms 色谱柱和DB-5ms 色谱柱,以每种溶剂在两个完全不同的色谱柱系统中的保留指数作为定性参数,建立KI 知识库。
在实验中发现,极性毛细管色谱柱VF-1301ms更适于36 种挥发性有机溶剂的定量分析。在极性毛细管色谱柱上,只有间二甲苯和对二甲苯保留指数、碎片离子完全相同,其余挥发性有机溶剂均能得到较好的分离和较高的响应。因此,选择极性毛细管色谱柱VF-1301ms 为定量色谱柱。
2.3 定性知识库的建立
2.3.1 双柱保留指数库的建立
保留指数(retention index,RI)或Kovats Index(KI)概念是由Kovats 在1958 年提出[8]。由于化妆品成分复杂,为了获得较好的分离分析效果,缩短分析时间,本项目采用程序升温保留指数来评价检测结果,计算公式如下:
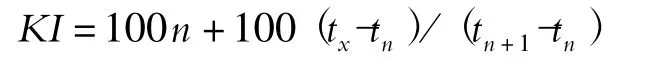
其中tx、tn和tn+1分别为被分析组分和碳原子数为n和n +1 的正烷烃流出峰的保留时间(min)(tn<tx<tn+1)。
化妆品中可能残留的溶剂种类复杂,分离度不佳或色谱条件发生改变会使组分的质谱图发生变化,使检索结果出现偏差[9]。另外,挥发性溶剂的相对分子质量小,电子轰击(EI)源轰击后碎片离子常常受到氮气、氧气、氩气等气体离子峰的干扰,导致质谱图复杂,检索NIST 谱库时结果不准确;挥发性溶剂的同分异构体多,如本文测定的正丁醇、异丁醇和乙酸丁酯、乙酸异丁酯等,仅使用NIST 谱库检索不能准确匹配这些异构体的谱图,如以样品中检出的异丁醇质谱图扣除背景后进行检索,检索结果表明正丁醇匹配指数更高。因此单纯检索NIST 谱库进行定性筛查结果不够准确。分析测定化妆品中可能含有的残留溶剂时,同时考虑色谱-质谱图和KI 值的匹配度,可大大提高鉴定结果的准确性。
2.3.2 双柱保留指数时间窗的建立
对知识库中包含的挥发性有机溶剂的保留指数进行精密度考察:(1)一天内6 次测定36 种挥发性有机溶剂在两个色谱系统中的保留指数,计算日内精密度;(2)连续3 天测定36 种挥发性有机溶剂在两个色谱系统中的保留指数,计算日间精密度。在极性色谱柱VF-1301ms 上,36 种挥发性有机溶剂的日内保留指数精密度和日间保留指数精密度均小于或等于0.1%;在非极性色谱柱DB-5ms 上,36 种挥发性有机溶剂的日内保留指数精密度和日间保留指数精密度也均小于或等于0.1%;考虑到不同仪器、色谱系统的差异,确定36 种挥发性有机溶剂的保留指数时间窗为1%。
2.4 色谱-质谱条件的选择
2.4.1 顶空条件的选择
2.4.1.1 平衡温度
将一定浓度的混合标准溶液分别在40、50、60、70、80 ℃温度下平衡15 min 后进行测定,得到36 种挥发性有机溶剂的峰面积和平衡温度关系。实验发现,在相同的平衡条件下,随着平衡温度的升高,峰面积随之增大,分析灵敏度也相应提高。但是由于平衡温度过高使顶空瓶的气密性变差,因此方法的精密度受到影响。所以在实际工作中往往在满足灵敏度的前提下选择较低的平衡温度。另外,考虑到化妆品成分的复杂性,平衡温度也不宜过高,因此选择60 ℃为顶空平衡温度。
2.4.1.2 平衡时间
将一定浓度的混合标准溶液在60 ℃下平衡10、15、20、30、40 min 后进行测定,得到36 种挥发性有机溶剂的峰面积和平衡时间的关系。实验发现,在相同的平衡温度下,平衡时间少于30 min 时,随着时间的增加,峰面积明显增加;当平衡时间超过30 min 后,峰面积不再增加,甚至略有下降。这可能是因为在30 min 后顶空瓶内已达到气液平衡,继续平衡导致顶空瓶的气密性变差,因此本实验选择30 min 为平衡时间。
2.4.2 色谱条件的选择
在进样口温度为150 ℃的条件下,考察了36 种挥发性有机溶剂在不同柱温、不同升温速率下的分离情况。实验发现,初始温度30 ℃(保持10 min),以5 ℃/min 升至100 ℃,30 ℃/min 升至220 ℃(保持5 min),36 种挥发性有机溶剂的色谱峰与杂质峰有良好的基线分离。最后的流出物质保留时间在20 min 左右,保留时间适宜。考察了分流比为10∶1、20∶1、50∶1、100∶1 时36 种挥发性有机溶剂的峰形,实验结果表明,36 种挥发性有机溶剂在分流比为50∶1 时得到最佳峰形和较高的灵敏度。实验中发现,36 种挥发性有机溶剂在极性毛细管色谱柱VF-1301ms 上的分离度和响应比在非极性毛细管色谱柱DB-5ms 上的结果略优,更适于定量分析,因此选择VF-1301ms 色谱柱作为定量色谱柱。对二甲苯和间二甲苯的保留时间、碎片离子相同,故按两种化合物的结果相加计算。
2.4.3 质谱条件的选择
采用气相色谱-质谱仪在m/z 30 ~200 范围内对36 种挥发性有机溶剂进行测定,发现主要干扰离子有m/z 28、32、40、44。采用SIM 检测模式可以排除这些离子,提高灵敏度。每种物质选择1 个定量离子,2 个定性离子。
在优化条件下,36 种挥发性有机溶剂在VF-1301ms 及DB-5ms 色谱柱上的总离子流图分别见图1 和图2。
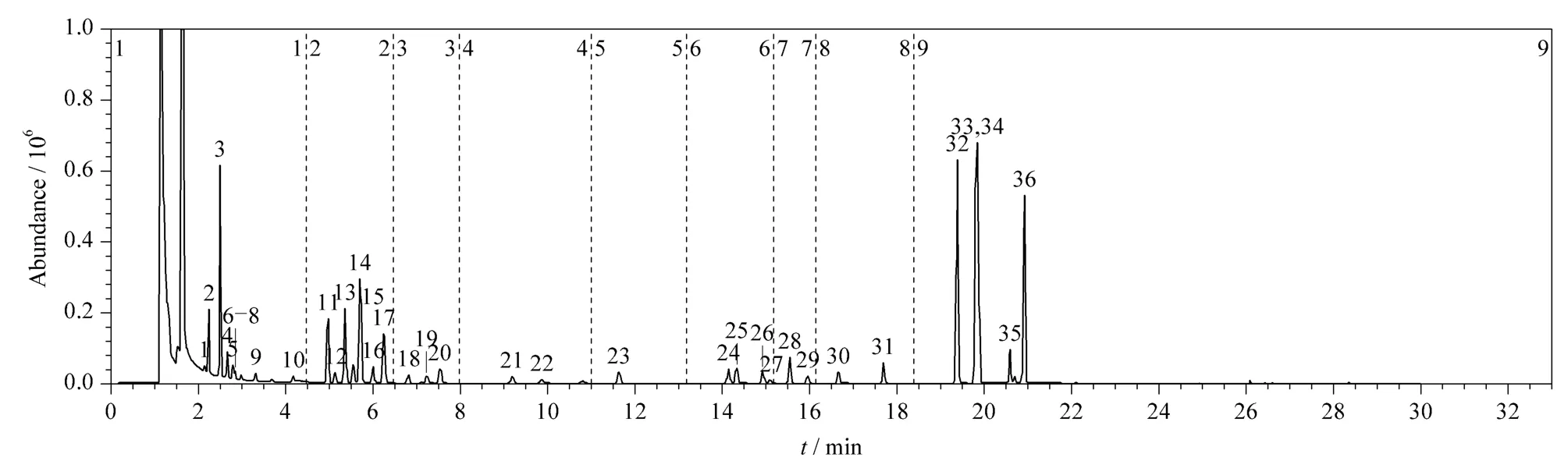
图1 36 种挥发性有机溶剂在VF-1301ms 极性柱上的总离子流图Fig.1 Total ion chromatogram of the 36 volatile organic solvents on polar column VF-1301ms

图2 36 种挥发性有机溶剂在DB-5ms 非极性柱上的总离子流图Fig.2 Total ion chromatogram of the 36 volatile organic solvents on nonpolar column DB-5ms
2.5 基质效应减小方法
2.5.1 稀释样品
稀释样品是减小样品基质效应的常用方法[10],但方法灵敏度会下降。通过实验发现,化妆水等黏度小的基质对环己烷等溶剂具有明显的基质增强效应;而乳液、膏霜状黏稠样品则对甲苯、乙基苯、间/对二甲苯、邻二甲苯有较强的基质抑制效应。但当称取1 g 样品,定容至100 mL 或以上时,不同黏度样品的加标回收率均在60% ~130% 之间。考虑到稀释比例过大会降低方法的灵敏度,故选择定容体积为100 mL。
2.5.2 利用盐析作用
在待测样品水溶液中加入NaCl,利用盐析作用降低挥发性有机溶剂在水溶液中的溶解度利于降低基质效应。实验表明,在10 mL 待测样品水溶液中加入1.0 g NaCl 可以达到明显的效果,加标回收率和灵敏度均能满足要求。
2.6 线性关系和检出限
由于不同挥发性有机溶剂在仪器上的响应值不同,在配制系列线性溶液时,各类挥发性有机溶剂按照不同的浓度进行配制。按1.5 节方法绘制工作曲线。根据现行的《化妆品卫生规范》中卫生化学检测方法总则的要求,气相色谱法的检出限(LOD)和定量限(LOQ)以3 倍空白噪声和10 倍空白噪声相对应的质量或浓度表示。
36 种挥发性溶剂的线性方程、相关系数、线性范围和在极性柱及非极性柱上的方法检出限、定量限见表5。
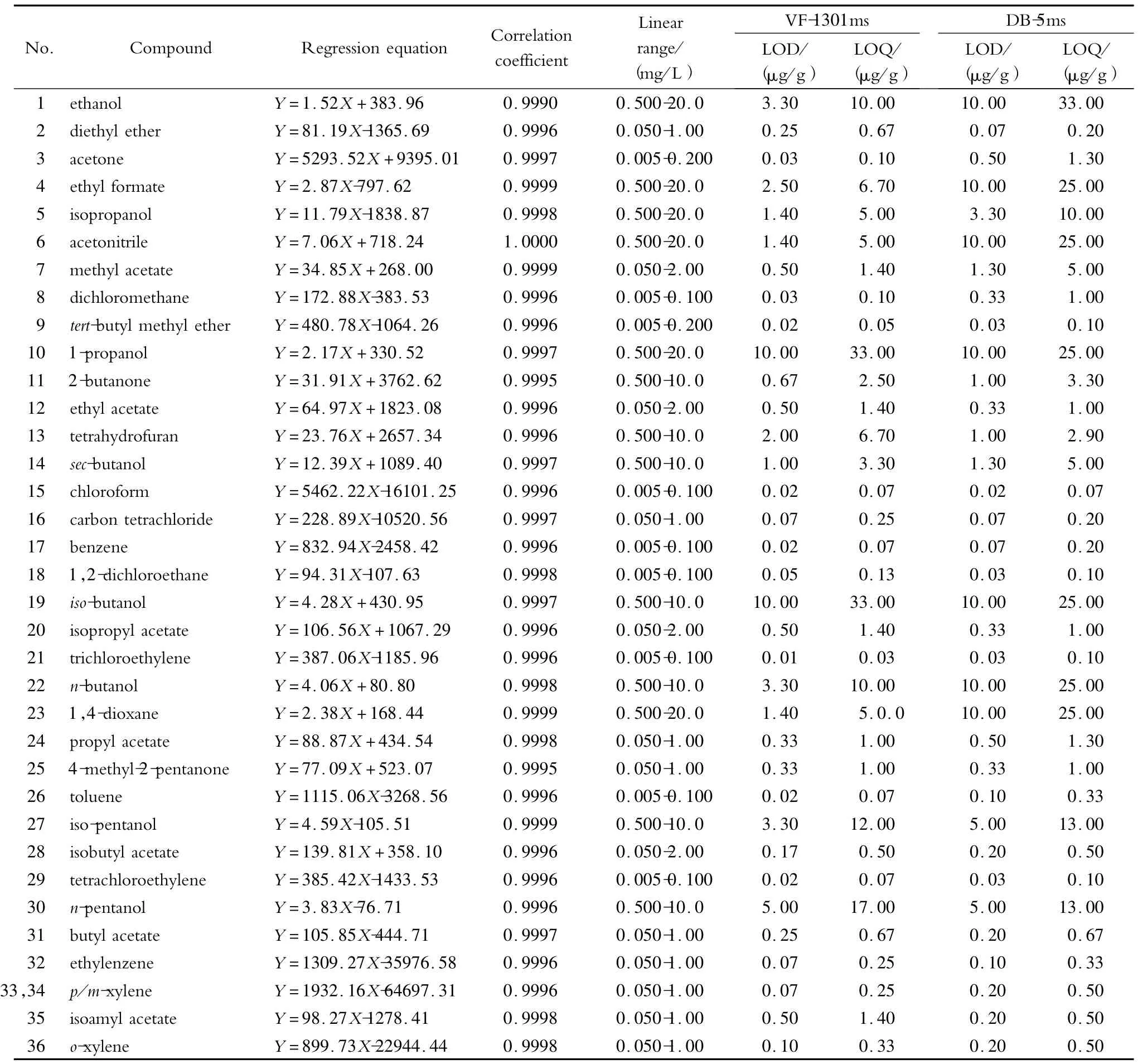
表5 36 种挥发性有机溶剂的标准曲线、相关系数、线性范围、检出限及定量限Table 5 Regression equations,correlation coefficients,linear ranges,limits of detection (LODs)and limits of quantification (LOQs)for the 36 volatile organic solvents
2.7 精密度和回收率
选择爽肤水、乳液、日霜3 种空白基质,分别加入低、中、高3 种浓度的混合标准溶液,进行加标回收试验和精密度试验。结果显示,36 种挥发性有机溶剂在爽肤水基质中高、中、低浓度的回收率在64.09% ~126.60%之间;36 种挥发性有机溶剂在乳液基质中高、中、低浓度的回收率在65.40% ~124.28%之间;36 种挥发性有机溶剂在日霜基质中高、中、低浓度的回收率在60.77% ~121.30%之间。3 种基质高中低3 个浓度回收率的RSD 均小于8.20%,满足定量分析方法的要求。
2.8 稳定性
挥发性有机溶剂的稳定性较差,因此考察了对照品溶液在24 h 内的稳定性。配制一定浓度的对照品溶液,分别在第0、2、4、8、12、24 h 测定。结果表明,四氯化碳、苯、三氯乙烯、甲苯、四氯乙烯、乙基苯、间/对二甲苯、邻二甲苯9 种挥发性有机溶剂在12 h 内稳定,其余挥发性有机溶剂在24 h 内均稳定。因此对照品溶液、样品溶液均需现用现配并立即测定。
3 样品测定
3.1 初筛知识库筛查样品
测定了7 类样品,分别为止汗喷雾、花露水、橄榄油、洗发水、爽肤水、眼霜及指甲油。在相同的色谱条件下测定正构烷烃的保留时间,计算出样品中可疑组分的双柱KI 值,将结果与表4 中的KI 值比较;同时用NIST 谱库检索可疑组分的质谱图,筛查可能存在的溶剂。筛查结果见表6。
在两个色谱系统中同时出现的色谱峰是样品中可能存在的挥发性有机溶剂:样品1 可能存在乙酸异戊酯;样品2 可能存在乙醇、乙酸乙酯;样品4 可能存在异丙醇;样品5 可能存在乙醇;样品7 可能存在二氯甲烷、正丁醇、乙酸异丁酯、乙酸异丙酯、异丙醇、乙酸乙酯、乙酸丙酯、乙酸丁酯;样品3 和样品6均未检出这几种有机溶剂。
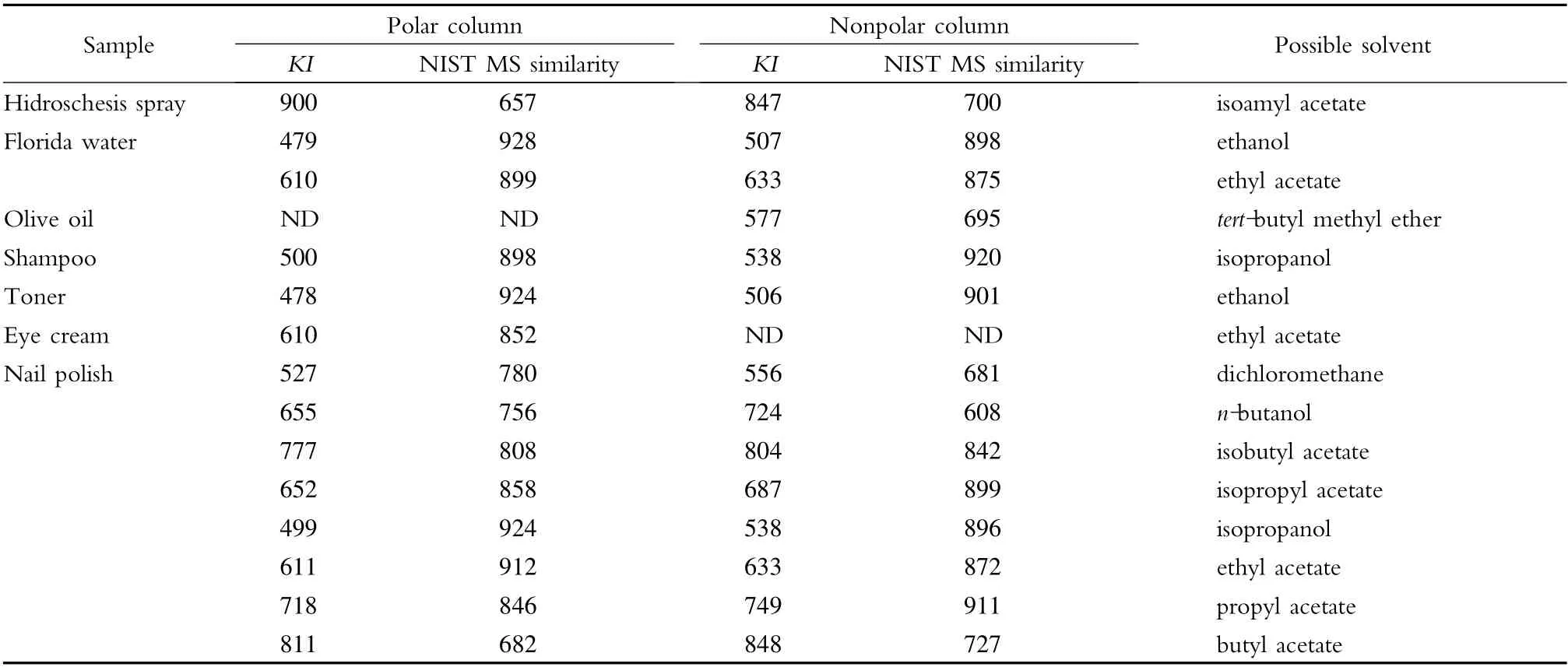
表6 7 类化妆品中挥发性有机溶剂的初筛结果Table 6 Prescreening results of the volatile organic solvents in seven cosmetics samples
3.2 确证及定量
配制一定浓度疑似存在组分的对照品溶液,选择VF-1301ms 毛细管色谱柱为分离柱,按照指定条件同时测定对照品溶液和样品溶液,比较对照品溶液与样品溶液中色谱峰的KI 值及离子丰度比,结果显示均一致,进一步说明样品中存在这些残留溶剂。对样品中存在的挥发性溶剂进行定量测定,结果表明样品1 中乙酸异戊酯含量为2.54 mg/kg;样品2中乙醇含量为496 749 mg/kg,乙酸乙酯含量为6.41 mg/kg;样品4 中异丙醇含量为20.26 mg/kg;样品5 中乙醇含量为40 255 mg/kg;样品7 中二氯甲烷、正丁醇、乙酸异丁酯、乙酸异丙酯、异丙醇、乙酸乙酯、乙酸丙酯和乙酸丁酯含量分别为5.06、3 340、117、2 079、42 172、228 995、72 506 和390 163 mg/kg。
4 结论
本文建立了36 种化妆品配方生产中常用的挥发性有机溶剂初筛知识库、确证知识库和定量测定方法,可以在未知化妆品生产工艺和配方的情况下快速、准确、全面的筛查、确证化妆品中的挥发性有机溶剂,并对存在的挥发性有机溶剂进行定量测定。
[1] Chen H Y,Xu Y H. Chinese Journal of Health Laboratory Technology (陈华宜,许瑛华. 中国卫生检验杂志),2009,19(7):1466
[2] Huang Q S. Modern Chemical Industry (黄秋森. 现代化工),2006,26(9):49
[3] Chen K,Yao Y Y. Environmental Monitoring in China (陈琨,姚媛艳. 中国环境监测),2011,27(2):59
[4] Department of Health of People’s Republic of China. Hygienic Standard for Cosmetics. 2007ed (中华人民共和国卫生部. 化妆品卫生规范. 2007 年版)
[5] Xu Y H,Zhu B H,Zhong X H,et al. Chinese Journal of Chromatography (许瑛华,朱炳辉,钟秀华,等. 色谱),2010,28(1):73
[6] Liu Y M,Ge N,Wang F,et al. Chinese Journal of Chromatography (刘永明,葛娜,王飞,等. 色谱),2012,30(8):782
[7] Liu Y,Hu C Q. Chinese Journal of Pharmaceutical Analysis (刘颖,胡昌勤. 药物分析杂志),2007,27(12):1938
[8] Zhao C X,Liang Y Z,Hu Q N,et al. Chinese Journal of Pharmaceutical Analysis (赵晨曦,梁逸曾,胡黔楠,等. 药物分析杂志),2005,33(5):715
[9] Liu P,Zhang Y,Lü Q T,et al. Food and Drug (刘朋,张莹,吕青涛,等. 食品与药品),2011,13(1):39
[10] Liu H W. GC Method and Its Application. 2nd ed. Beijing:Chemical Industry Press (刘虎威. 气相色谱方法及应用. 2版. 北京:化学工业出版社),2007
