肉桂多糖的提取纯化及体外抗氧化和降血糖活性分析
2024-04-01张慧慧刘会平马笑笑
张慧慧,李 灿,刘会平,马笑笑,张 欣,王 兵,刘 盈
(天津科技大学食品科学与工程学院,天津 300457)
肉桂(Cinnamomum cassia)是樟科樟属植物肉桂的干燥树皮,既可药用也常被用做食品香料,调味品等,国家卫生部将其划分为药食同源物品,在我国广泛分布于广西等热带或亚热带地区[1]。肉桂中含有多种活性成分,如肉桂醛、萜类物质、苯丙素、多糖等,因此具有多种生物活性[2]。已有大量研究表明,肉桂具有抗氧化、抑菌抗炎、抗凝血、抗糖尿病等功能作用[3-7]。
目前已有的与各种肉桂多糖相关的研究主要如下:Al-Ajalein 等[8]用微波辅助的方法从肉桂树皮中提取到一种低分子量果胶多糖,主要由葡萄糖、阿拉伯糖、半乳糖、半乳糖醛酸组成,研究发现其具有抗色素沉着过度和酪氨酸酶抑制作用。李胜男等[9]从肉桂水提物中分离纯化出分子量为3630 Da 的中性多糖,主要由葡萄糖组成,具有良好的DPPH·和ABTS+·清除能力。张铭儒等[10]制备到主要由甘露糖、半乳糖醛酸、葡萄糖、半乳糖、阿拉伯糖组成的肉桂多糖,并发现其可能通过调节糖脂代谢而显示出对糖尿病小鼠降血糖作用。此外,于峰等[11]也发现肉桂多糖对四氧嘧啶诱发的实验性糖尿病小鼠有显著的降糖效果。
但不同来源的肉桂其活性物质的结构性质存在部分差异[12],关于某种特定资源肉桂多糖的研究鲜有报道。“防城肉桂”是广西防城港市特产,中国地理标志证明商标,资源丰富,品质独特,故以防城港肉桂为原料,采用水提醇沉法提取肉桂多糖,探究其体外抗氧化活性及降血糖活性,以期为防城港肉桂资源的综合利用提供理论基础。
1 材料与方法
1.1 材料与仪器
肉桂 产自广西省防城港;无水乙醇、正丁醇天津市江天化工技术股份有限公司;三氯甲烷 天津市化学试剂厂;2,1-二苯基-2-三硝基肼(DPPH)、2,2'-联氨-双(3-乙基苯并噻唑啉-6-磺酸)二铵盐(ABTS)、硫酸亚铁、水杨酸、过氧化氢、铁氰化钾、氯化铁等 麦克林生化科技有限公司;Sephadex G-200、无水葡萄糖、AB-8 大孔吸附树脂、葡聚糖标准品 北京索莱宝科技有限公司;阿卡波糖、α-淀粉酶(5 U/mg)、α-葡萄糖苷酶(7.5 U/mg) 上海源叶生物科技有限公司;所用试剂均为分析纯。
SE-2000 高速粉碎机 圣象电器有限公司;ESJ205-4 电子天平 Mettler Toledo;DF-101S 集热式恒温加热磁力搅拌器 杭州旌斐仪器科技有限公司;RE-52A 旋转蒸发仪 上海亚荣生化仪器厂;H1850R 高速冷冻离心机 湖南湘仪离心机仪器有限公司;BS-100A 自动部分收集器 上海沪西分西仪器有限公司;FD-1A-50 真空冷冻干燥机 上海比郎仪器制造有限公司;Agilent 1260 高效液相色谱仪美国安捷伦科技公司;VECTOR-220 傅里叶红外光谱仪 德国布鲁克公司;Multiskan GO 酶标仪美国Thermo Fisher 公司。
1.2 实验方法
1.2.1 肉桂多糖的提取 肉桂用高速粉碎机粉碎,将粉末用95%乙醇搅拌5 h 进行脱脂脱色[13],结束后滤去乙醇溶液,沉淀于55 ℃彻底干燥。采用传统水提醇沉法提取肉桂多糖。热水浸提2 次后合并滤液,真空旋转蒸发至原四分之一体积,随后边搅拌边加入无水乙醇至溶液浓度为60%,4 ℃沉淀18 h。结束后,离心(4000 r/min)15 min 分离沉淀,散至无乙醇味,加入蒸馏水复溶,冷冻后真空干燥。肉桂粗多糖得率计算公式如下:
1.2.2 单因素实验 为探究不同提取条件对肉桂多糖得率的影响,选定提取温度90 ℃,时间2 h,液料比20:1(mL/g),提取1 次,醇沉浓度60%的条件,考察浸提温度(60、70、80、90、100 ℃),浸提时间(1、2、2.5、3、3.5 h),液料比(10:1、15:1、20:1、25:1、30:1 mL/g),浸提次数(1、2、3、4 次)对粗多糖得率的影响。
1.2.3 响应面优化试验 根据单因素实验结果,固定浸提次数2 次,选择三个变量:浸提温度、浸提时间、液料比,运用Design Expert 13 软件进行三因素三水平的Box-Behnken 试验设计。试验因素与水平见表1。
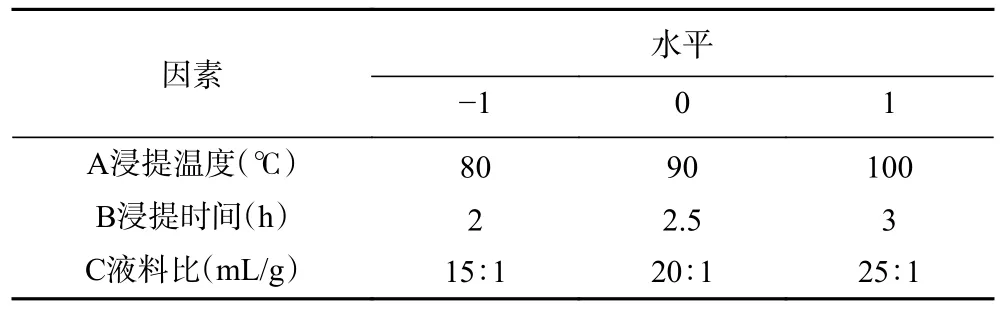
表1 响应面试验的因素与水平Table 1 Factors and levels of response surface experiment
1.2.4 肉桂多糖的纯化 通过Sevag 法(V三氯甲烷:V正丁醇=4:1)去除蛋白质,取Sevag 试剂与4 倍体积粗多糖溶液混合并振荡,离心,直至不出现蛋白层,重复约7 次。然后将除蛋白后的粗多糖溶液通过预处理后的AB-8 大孔吸附树脂层析柱以恒流泵30 r/min的流速进行脱色[14],将脱色成功的溶液浓缩,蒸馏水透析72 h,冷冻干燥后获得粗多糖。配制20 mg/mL 的多糖溶液,过0.22 μm 滤膜后加样至Sephadex G-200 层析柱,以蒸馏水作洗脱剂,流速为3 min/mL,每管收集2 mL。用苯酚-硫酸法[15]测定每管洗脱液中多糖含量,绘制出洗脱曲线,其中洗脱曲线的横坐标为收集的试管号,纵坐标为490 nm 处洗脱液的吸光度值。收集曲线峰管中的洗脱液,冷冻干燥得纯化的肉桂多糖,命名为CCP。
1.2.5 CCP 含量测定 采用苯酚-硫酸法[15]测定CCP 的总糖含量,其中标准品是无水葡萄糖,标准曲线为葡萄糖含量-吸光度值图。具体操作如下:
分别取0.1 mg/mL 葡萄糖标准溶液0、0.2、0.4、0.6、0.8、1 mL 于玻璃试管,补充蒸馏水至总体积为1 mL,随后各加入1 mL 6%苯酚溶液和5 mL浓硫酸,振荡混匀后室温静置10 min,沸水浴15 min,降温后测定各试管溶液在490 nm 处的吸光度。将CCP 配制成0.1 mg/mL 的溶液,取1 mL 按照上述方法处理后测定溶液的吸光度值。根据标准曲线计算CCP 的总糖含量,计算公式如下:
式中,X 为样品组吸光度值代入标准曲线后所得葡萄糖含量(mg);m 为样品质量(mg)。
1.2.6 CCP 相对分子质量 将葡聚糖标准品(T10、T40、T70、T110、T500、T2000)及CCP 各自溶解在蒸馏水中(1 mg/mL)并通过0.22 μm 滤膜。以超纯水作流动相,采用高效液相色谱仪中示差检测器和凝胶色谱柱对所备样品进行检测,运行参数为柱温30 ℃,进样体积20 μL,洗脱速度0.6 mL/min。根据各标准品保留时间-分子量lg 值所得的标准曲线计算CCP 的相对分子质量,计算公式如下:
式中,y 为样品保留时间代入标准曲线后所得的值。
1.2.7 CCP 的傅里叶红外光谱分析 称取1 mg CCP和150 mg 干燥KBr 置于研钵并快速研磨成粉末,在12 MPa 压力下压成均匀透光的薄片。通过傅里叶红外光谱仪在4000~400 cm-1范围以4 cm-1的分辨率识别CCP 的特征吸收峰,扫描16 次。
1.2.8 CCP 的1H 核磁共振检测 称取50 mg CCP,用0.5 mL D2O 充分溶解并移入核磁管中,利用DPX-400 核磁共振波谱仪记录1H 谱。
1.2.9 刚果红分析 参考Ma 等[16]的方法,配制浓度为0.5 mg/mL CCP 溶液、50 μmol/L 刚果红溶液和1 mol/L NaOH 溶液。取2 mL CCP 溶液和等体积刚果红溶液,再加入不同体积的蒸馏水和NaOH溶液,最终使NaOH 浓度分别为0、0.05、0.1、0.15、0.2、0.3、0.4 mol/L,室温下反应10 min 后,使用紫外分光光度计在400~600 nm 范围内测定不同梯度溶液的最大吸收波长。
1.2.10 热重分析 称取5 mg CCP 样品置于坩埚中,设置初始温度为30 ℃,结束温度为600 ℃,以10 ℃/min 的加热速度进行热力学测试。
1.2.11 CCP 的体外抗氧化活性测定 实验参考的方法均略有改动,具体方案如下。
1.2.11.1 DPPH 自由基清除能力测定 方法参考Deng 等[17],配制0.2 mmol/L DPPH 无水乙醇溶液,不同浓度CCP 溶液(0.05、0.1、0.5、1.0、2.0、3.0、4.0、6.0 mg/mL),取1 mL CCP 溶液与2 倍的DPPH无水乙醇溶液混匀,37 ℃黑暗反应30 min,检测溶液在517 nm 处的吸光度值。同时,以VC为阳性对照。
式中,A1是CCP 溶液组吸光度值;A2是DPPH无水乙醇换成无水乙醇;A0是多糖溶液换成蒸馏水。
1.2.11.2 ABTS+自由基清除能力测定 方法参考Thambiraj 等[18],配制ABTS+·工作液(等体积混合7 mmol/L ABTS 和2.45 mmol/L 过硫酸钾,用蒸馏水将混合液在734 nm 处的吸光度值调整为0.7±0.02)。取0.4 mL 不同浓度的CCP 溶液(同1.2.11.1)与4 mL 工作液混匀,黑暗反应5 min,检测溶液在734 nm 处的吸光度值。同样以VC为阳性对照。
式中,A1是CCP 溶液组吸光度值;A2是ABTS+·工作液换成蒸馏水;A0是多糖溶液换成蒸馏水。1.2.11.3 羟自由基清除能力测定 方法参考Zhang等[19],准备6 mmol/L 的硫酸亚铁溶液和水杨酸·无水乙醇溶液、0.1%的过氧化氢溶液、不同质量浓度的CCP 溶液(同1.2.11.1)。取1 mL 多糖溶液依次加入等体积的三种试剂并充分混合,37 ℃反应30 min 后,检测混合液在510 nm 处的吸光度值。以VC为阳性对照。
式中,A1是CCP 溶液组吸光度值;A2是过氧化氢溶液换成蒸馏水;A0是多糖溶液换成蒸馏水。
1.2.11.4 总还原力测定 方法参考Chen 等[20],配制0.2 mol/L pH6.6 的磷酸缓冲液、1%铁氰化钾、10%三氯乙酸、0.1%氯化铁、不同质量浓度的CCP溶液(同1.2.11.1)。将1 mL 多糖溶液、2.5 mL 的磷酸缓冲液和铁氰化钾溶液混匀,50 ℃水浴20 min。冷却后再加2.5 mL 三氯乙酸,离心后取上清2.5 mL,加入等体积蒸馏水和0.5 mL 氯化铁溶液,检测混合溶液在700 nm 处的吸光度值,最终以吸光度值大小表示总还原力的强弱。以VC为阳性对照。
1.2.12 CCP 的α-淀粉酶抑制活性 方法参考Cao等[21],但有改动。用0.1 mol/L 的PBS 配制不同质量浓度(0.05、0.1、0.25、0.5、0.75、1.0、1.5 mg/ mL)的肉桂多糖溶液,酶活力为50 U/mL 的α-淀粉酶溶液以及1%的可溶性淀粉溶液。取多糖样品和α-淀粉酶溶液各30 µL,37 ℃反应10 min,再加入同体积可溶性淀粉溶液,沸水浴15 min,迅速冷却后向其中加入50 µL DNS,然后95 ℃加热5 min,冷却后加入500 µL 蒸馏水,检测溶液在540 nm 处的吸光度值,以阿卡波糖为阳性对照。
式中,A1是CCP 溶液组吸光度值;A2是将α-淀粉酶溶液换成PBS;A3是将多糖溶液换成PBS。
1.2.13 CCP 的α-葡萄糖苷酶抑制活性 方法参考Xu 等[22],但有改动。用0.1 mol/L 的PBS 配制1.2.12中的多糖溶液,1 U/mL 的α-葡萄糖苷酶溶液、5 mmol/L 的4-硝基苯基-β-D-吡喃葡萄糖苷(PNPG)。取多糖溶液与α-葡萄糖苷酶各40 µL 混匀,37 ℃孵育10 min,然后加入40 µL PNPG,同温度再孵育5 min,再加入100 µL Na2CO3溶液(1 mol/L)终止反应并显色,检测溶液在405 nm 处的吸光度值。阳性对照为阿卡波糖。
式中,A1是CCP 溶液组吸光度值;A2是将α-葡萄糖苷酶溶液换成PBS;A3是将多糖溶液换成PBS。
1.3 数据处理
实验设三次平行,数据以平均值±标准差表示。运用Design Expert 13、Origin Pro 2021、Graph Pad Prism 8 软件对实验数据进行响应面分析与统计学分析。
2 结果与分析
2.1 单因素实验结果
肉桂多糖得率随提取温度的升高而升高,温度达到90 ℃时,得率为3.22%,温度继续升高到100 ℃时,多糖的溶出量基本稳定,多糖得率并未再提升。因此选择80、90、100 ℃三个温度点参与响应面设计。
浸提时间高于2.5 h 之后,多糖得率的升高速率趋于平缓,可见长时间的浸提不会提升得率,可能是后期出现了杂质的溶出。考虑到提取效率,选取2、2.5、3 h 继续响应面试验。
溶剂在一定范围内增加可能使细胞内外浓度差增大,从而使多糖更多地溶出。液料比在20:1 mL/g之后多糖得率开始下降,可能是随着溶剂量的增加,此时多糖成分提取已比较充分,升高的压力差导致其它溶质溶出[23]。因此选择15:1、20:1、25:1 mL/g作为响应面试验水平。
从图1 可以看到,当浸提次数超过2 时对肉桂多糖得率的影响并不明显,考虑总体提取时间、效率等因素,确定提取次数为2 次。

图1 不同单因素条件下的肉桂多糖提取效果Fig.1 Extraction efficiency of Cinnamomum cassia polysaccharide with different single factor conditions
2.2 响应面试验结果
2.2.1 试验设计与结果 表2 是响应面试验设计方案及最终结果,对此结果进行多元拟合分析,获得预测模型方程:Y=3.32+0.1425A+0.1988B+0.1213C+0.0375AB+0.0175AC+0.0400BC-0.2280A2-0.2755B2-0.3455C2。

表2 响应面试验设计及结果Table 2 Response surface experiment design and results
2.2.2 各因素交互作用分析 方差分析由表3 可见,该模型差异性(P<0.0001)极显著,失拟项(P>0.05)不显著,说明模型拟合较好;同时根据P值可知[24],回归模型的一次项影响均极显著(P<0.0001),交互项AC 影响不显著(P>0.05),AB、BC 影响显著(P<0.05),二次项均影响极显著(P<0.0001)。由F值大小可知,试验中各因素对得率的影响顺序为(B)时间>(A)温度>(C)液料比。
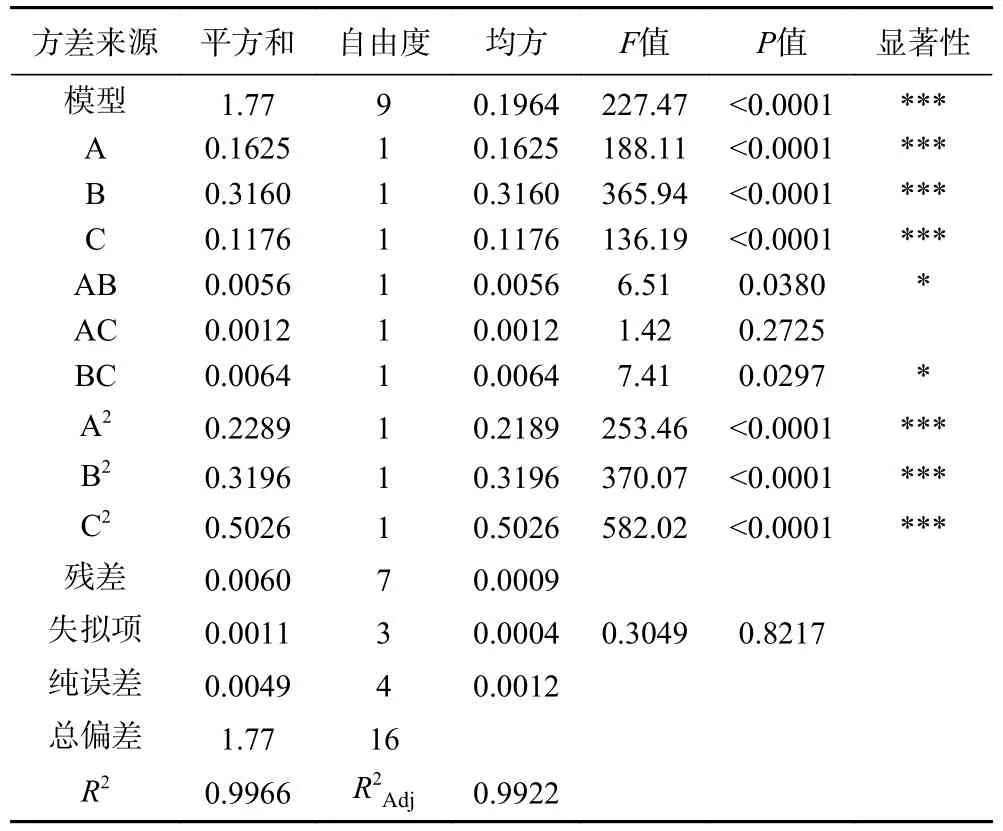
表3 回归模型方差分析结果Table 3 Variance analysis for response surface quadratic model
由图2 可以看出,响应面图表面相对陡峭,等高线轮廓形状为椭圆形,相对致密,说明这些提取因素共同影响着肉桂多糖得率[25],但可以看出图2B、2b 中这些形状更为明显,即浸提时间和液料比的交互作用对多糖得率的影响最为显著,这也与方差分析中交互项的P值对应;所有响应面均开口向下,表明回归模型具有极值点[25],此时理论提取工艺为温度91.98 ℃,时间2.67 h,液料比20.67:1(mL/g),预测得率为3.39%。综合操作等实际因素,调整提取条件为温度90 ℃,时间2.5 h,液料比20:1(mL/g),以此条件进行3 次实验的平均得率为3.22%,与理论值的相对误差为0.17%。结果表明了模型的适用性及条件优化的成功[24]。
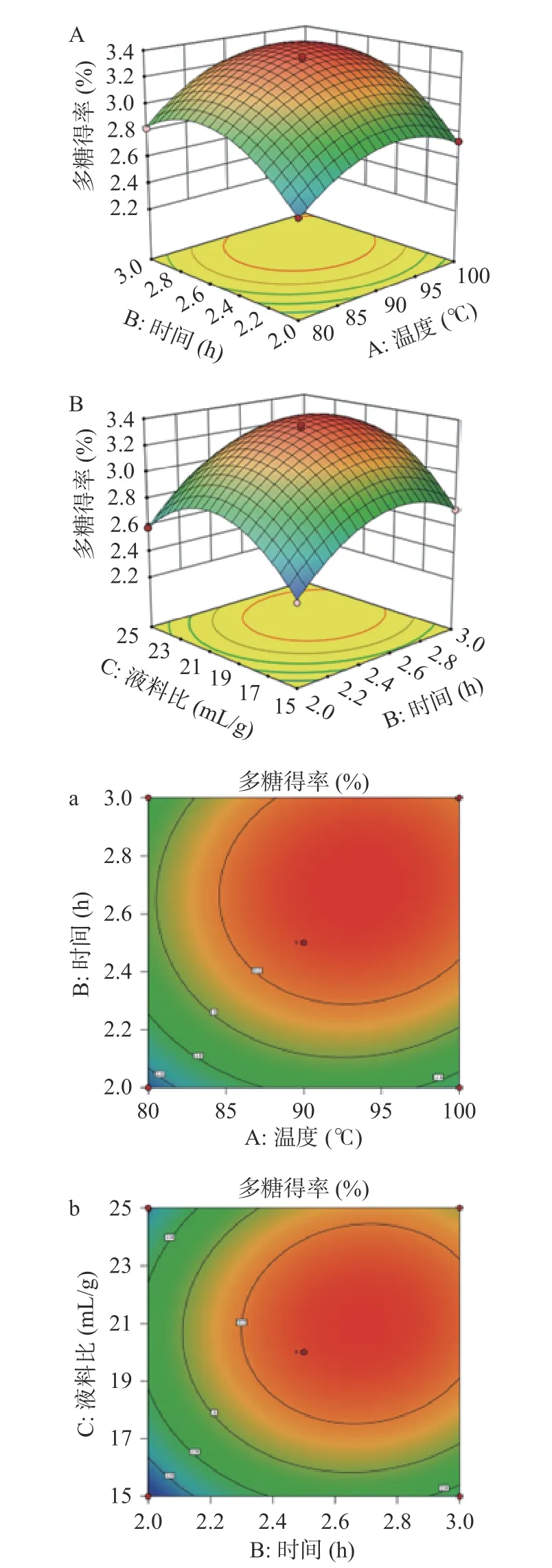
图2 显著性交互作用因素对多糖得率影响的响应面图(A、B)和等高线图(a、b)Fig.2 Response surface diagram (A, B) and contour diagram(a, b) of the influence of significant interaction factors on polysaccharide yield
2.3 肉桂多糖的纯化
2.3.1 肉桂多糖的纯化 由图3 可知,肉桂多糖通过Sephadex G-200 层析柱纯化后主要得到一个组分,收集洗脱曲线9、10 管洗脱液,冷冻干燥后备用。
2.3.2 CCP 的多糖含量 由葡萄糖标准品获得的标准回归方程为y=5.5064x+0.1268,R2=0.9967,通过计算得出CCP 总糖含量为90.11%±1.24%。
2.3.3 CCP 相对分子质量 由葡聚糖标准品得到标准回归方程为y=-0.3555x+9.3736,R2=0.9906。经计算CCP 的相对分子质量为1.95×106Da。
2.4 傅里叶红外光谱分析
CCP 的红外光谱图(图4)如下,3416 cm-1附近出现的强吸收峰归因于O-H 的伸缩振动,表明分子间或分子内氢键的存在[26]。2924 cm-1附近的吸收峰归因于C-H 的伸缩振动[27]。1739 cm-1处的吸收峰是酯羰基的典型特征[28]。1631 cm-1处是C=O 吸收峰[29]。1420 cm-1处的吸收峰归因于C-O 基团的伸缩振动[30]。1200~1000 cm-1之间的三个吸收峰表明存在吡喃糖环[31]。927 cm-1和851 cm-1左右的峰表明多糖中可能存在β-型糖苷键和α-型糖苷键[32]。

图4 CCP 的傅里叶红外光谱图Fig.4 Infrared spectroscopy of CCP
2.5 CCP 的1H 谱分析结果
在1H 谱图(图5)中,未被标注的最强信号峰属于D2O 溶剂峰;δH4.3~5.9 ppm 是异头质子信号区,通常异常区域内化学位移大于5.0 ppm 的异头质子对应α构型,大于4.3 ppm 的异头质子对应β构型[30,33],由1H 谱图结果可知,CCP 存在α和β型糖残基,这与红外光谱分析结果一致;此外,δH3.5~4.0 ppm 区域是糖环的质子信号[19]。

图5 CCP 的1H 核磁共振谱图Fig.5 1H nuclear magnetic resonance spectrogram of CCP
2.6 CCP 的刚果红分析
刚果红染料能与含有三股螺旋构象的多糖形成络合物,它的存在会使最大吸收波长与刚果红溶液相比发生红移,但随着NaOH 浓度增大,络合物结构被破坏,溶液最大吸收波长会下降[34]。图6 显示了两组溶液在扫描范围内整体吸光度值的变化;图7 可以看到,在不同浓度NaOH 溶液中,刚果红和肉桂多糖混合液的最大吸收波长并未发生红移后再下降的现象,表明CCP 可能不含三股螺旋结构[19]。
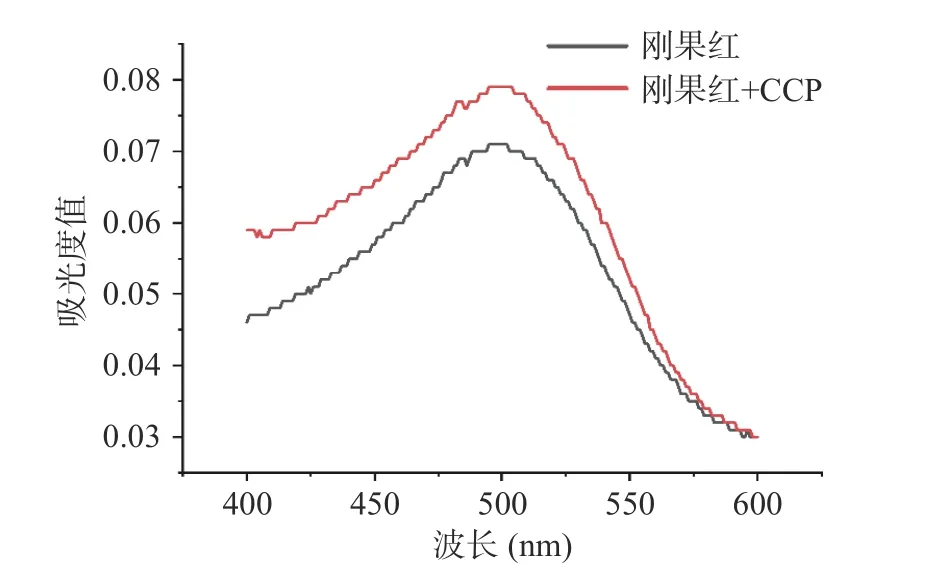
图6 刚果红和刚果红+CCP 溶液(不含NaOH)的吸光度值Fig.6 Absorbance value of Congo red and Congo red+CCP solution (without NaOH)
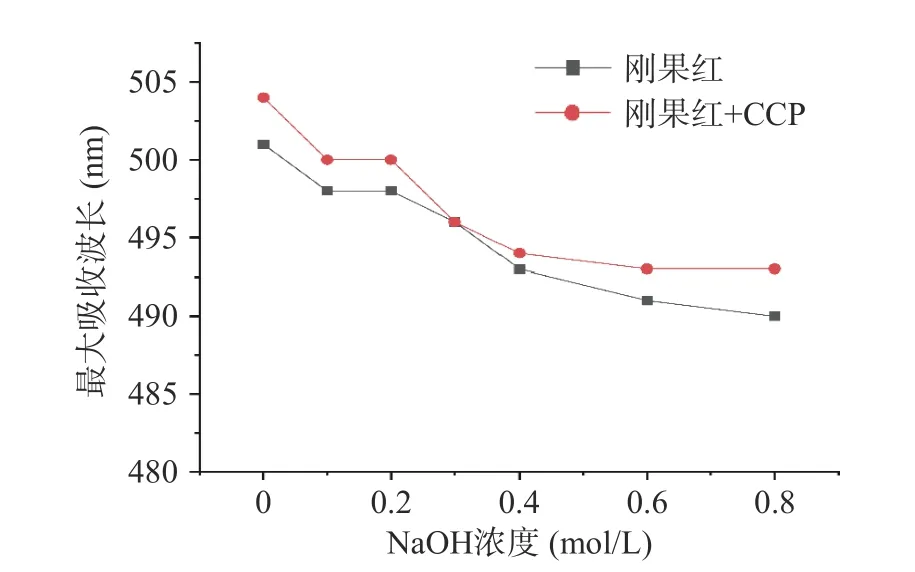
图7 刚果红和刚果红+CCP 溶液的最大吸收波长示意图Fig.7 Trends of maximum absorption wavelengths of Congo red and Congo red+CCP solution
2.7 CCP 的热重分析
CCP 的热失重过程分三个阶段进行(图8)。第一阶段发生在60 ℃左右,失去其重量的6.16%,归因于多糖失去吸附水;第二阶段发生的温度范围是240~360 ℃,失去其重量的74.11%,表明多糖在这一温度范围内发生了强烈的分解反应;最后,在400~600 ℃温度范围内,多糖重量变化趋于平缓,为缓慢碳化阶段,大部分变成灰分和无机成分。
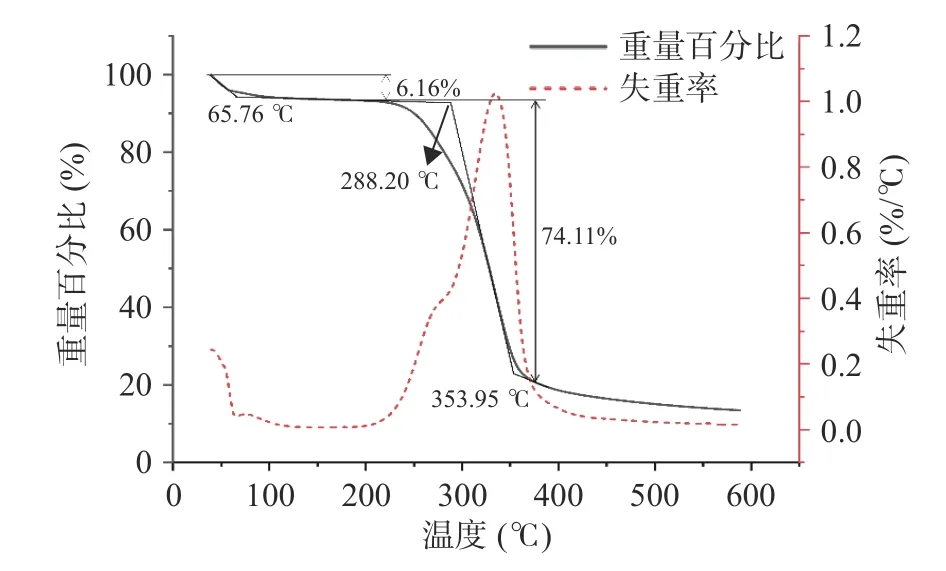
图8 CCP 的热重分析Fig.8 Thermogravimetric analysis of CCP
2.8 CCP 的体外抗氧化活性
许多因素会影响抗氧化活性,单一的抗氧化模型无法完全反映样品各方面的抗氧化能力[35],因此选择多种模型评估CCP 的抗氧化能力。
2.8.1 DPPH 自由基清除能力 DPPH 自由基常被用来评估还原物质,是研究活性物质对自由基清除能力的有用试剂[36]。由图9 可以看出,在0.05~6 mg/mL浓度范围内,CCP 对DPPH·的清除能力逐渐增强,但后面增加缓慢。在6 mg/mL 时的清除率达到81.32%,肉桂多糖和VC清除DPPH·的IC50值分别为0.191、0.008 mg/mL。
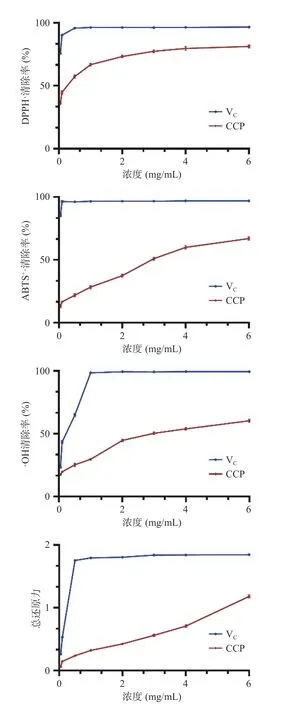
图9 CCP 的抗氧化能力Fig.9 Antioxidant capacity of CCP
2.8.2 ABTS+自由基清除能力 ABTS+自由基清除实验是确定供氢和断链抗氧化剂活性的有效方法[36]。如图9 所示,在实验剂量范围内,ABTS+·清除率与肉桂多糖浓度呈正相关,肉桂多糖和VC清除ABTS+·的IC50值分别为2.835、0.0002 mg/mL。
2.8.3 羟自由基清除能力 羟自由基是在人体内产生且损害生物体的高活性物质,因此,测试对这些生物相关自由基的清除能力是很有必要的[37]。如图9所示,肉桂多糖也具有一定的羟自由基清除能力,最大浓度6 mg/mL 时的清除率为60.23%,肉桂多糖和VC清除羟自由基的IC50值分别为3.221、0.149 mg/mL。
2.8.4 总还原能力 该实验是测试活性物质对三价铁的还原能力,被广泛用于分析植物提取物的抗氧化活性[38]。实验结果中吸光度值直接反映总还原力的大小,如图9 所示,肉桂多糖的总还原力随肉桂多糖的质量浓度升高而增大,在6 mg/mL 时的最大吸光度值约为1.18,此时抗坏血酸的吸光度值约为1.85。
在4 种抗氧化试验中,VC浓度达到1 mg/mL 时的清除率都能达到95%以上,而CCP 对DPPH·的清除效果最好。此外对比发现CCP 对DPPH·,ABTS+·的清除率没有李胜男等[9]测试的结果高,有研究显示小分子量多糖比大分子量的有更强的抗氧化活性[22],而本研究分离到的肉桂多糖分子量远大于李胜男等[9]的(3630 Da),可能是此原因导致两者抗氧化能力不同。但与同样的高分子量羊栖菜多糖相比,肉桂多糖的部分自由基(DPPH·、·OH)清除能力更高,这可能与多糖的其它结构有关[20]。
2.9 CCP 的体外降血糖活性
2.9.1α-淀粉酶抑制活性α-淀粉酶的活性被抑制后可以延缓碳水化合物的消化,降低葡萄糖的吸收率,从而减缓餐后血糖的升高速度[39]。试验结果见图10,在多糖最大浓度1.5 mg/mL 时,抑制率为79.73%。肉桂多糖、阿卡波糖的IC50值对应为0.189、0.003 mg/mL。

图10 CCP 的α-淀粉酶抑制曲线Fig.10 α-Amylase inhibition curve of CCP
2.9.2α-葡萄糖苷酶抑制活性α-葡萄糖苷酶参与人体糖代谢,抑制其活性可以延缓肠道中碳水化合物的吸收,从而达到降糖效果[40]。由图11 所示,同浓度范围内,CCP 对α-葡萄糖苷酶的抑制率不如α-淀粉酶,但也可以达到67.13%,肉桂多糖、阿卡波糖的IC50值分别为0.340、0.004 mg/mL。
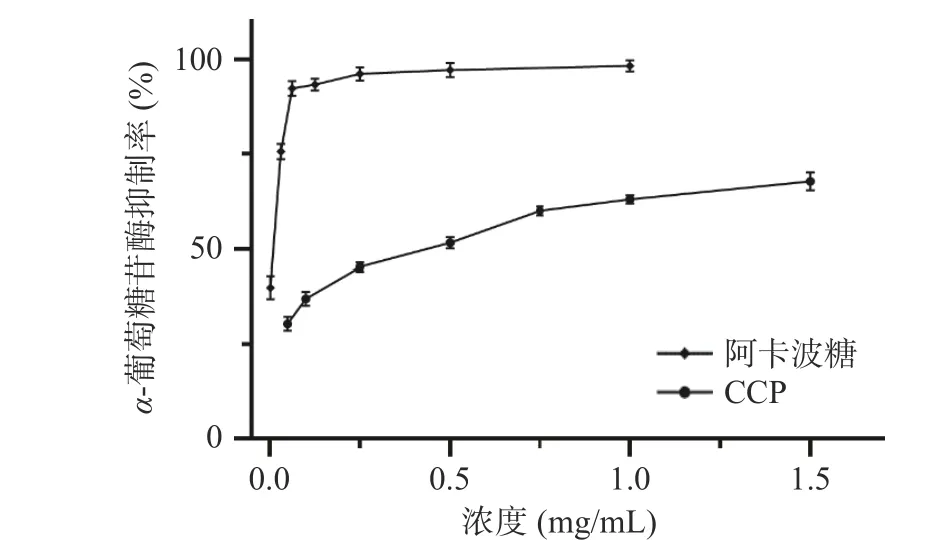
图11 CCP 的α-葡萄糖苷酶抑制曲线Fig.11 α-Glucosidase inhibition curve of CCP
肉桂多糖对两种酶显示出较好的抑制活性,但远低于阿卡波糖,然而与其它研究相比,肉桂多糖对两种酶活力仍具有更好的抑制能力[22,41]。
3 结论
肉桂作为药食同源物品,其内各种活性成分得到了广泛的关注。本研究优化了肉桂多糖的提取工艺,最佳工艺平均得率为3.22%。多糖经脱蛋白、脱色、葡聚糖凝胶纯化后,总糖含量为90.11%±1.24%,相对分子质量为1.95×106Da。CCP 部分结构测定显示其可能存在α-型和β-型糖苷键以及吡喃糖环,不含有三股螺旋结构。热重结果显示其具有较好的热稳定性,230 ℃以下稳定。在活性实验中,CCP 显示出较好的自由基清除能力和较高的总还原力;此外,CCP 也通过α-淀粉酶和α-葡萄糖苷酶抑制试验而显示出具有良好的降血糖活性。
由于本研究获得的多糖分子量较大,其结构特征可能更为复杂,因此研究主要集中于肉桂多糖的提取纯化、部分一级结构分析及活性探究,在未来的研究中将借助更多方法及分析仪器,表征肉桂多糖一级结构,深入探讨其与生物活性之间的关系。
本研究从防城肉桂中提取到一种新的高分子量多糖,该多糖在功能性食品或医药应用方面具有潜在的抗氧化及降血糖特性,上述结果可能为不同来源肉桂多糖的结构及活性研究提供参考,同时也为肉桂在食品药品等领域的开发利用提供理论依据。
© The Author(s) 2024.This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by-nc-nd/4.0/).
