Li/SF6 燃烧反应路径及机理研究
2024-01-13温伯尧王起源孙成珍骆政园白博峰
温伯尧 ,王起源 ,孙成珍 ,宗 潇 ,骆政园 ,白博峰 *
(1.西安交通大学 动力工程多相流国家重点实验室,陕西 西安,710049;2.中国船舶集团有限公司 第705 研究所,陕西 西安,710077)
0 引言
由碱或碱土金属(如Li、Al、Mg 等)和强氧化剂组成的金属燃料系统相比于碳氢燃料系统,具有能量密度高、体积小、低噪声等优点,在能源动力领域具有非常显著的优越性[1-3]。其中,由于Li与SF6是比较理想的金属燃料和氧化剂,具有反应产热高、反应过程稳定、反应物易存放、无气态产物排放、安全以及无噪声等优点[4-6],因此Li/SF6通常被用作热源,可与不同热机搭配构成闭式循环热动力系统。Li/SF6燃烧反应放热剧烈,产物十分复杂,反应机理涉及化学反应、相变、高温气体裂解、多相流动和湍流等复杂物理化学过程。揭示Li/SF6燃烧微观反应机理对于构建金属燃料反应动力学模型、实现燃烧过程高效组织具有十分重要的指导和实际意义。
Li/SF6作为最具代表性的金属燃料系统之一,国内外学者对其进行了大量研究,主要包括反应机理研究、实验观测及数值仿真等。20 世纪60 年代,荷兰研究人员发表了多组分在不互溶相中的热力学模型和热化学数据、Li/SF6浸没燃烧的多流体湍动模型[7];1992 年美国爱荷华大学的Lyu等[8-9]提出了一个基于守恒标量方法和界面分析的Li/SF6燃烧预测模型。我国相关研究起步较晚,朱强[10-11]计算了Li/SF6反应产物Li2S 的比热、焓和熵等热力学数据,并将结果拟合成多项式,这些数据在Li/SF6闭式循环动力系统研究中得到了应用;张文群等[12-14]通过建立Li/SF6气液浸没燃烧反应相平衡计算的非线性约束优化方法,计算了反应的平衡产物、温度和密度等,给出了Li 初始温度的影响,并利用Gibbs 自由能最小法研究了Li/SF6气液浸没燃烧反应中混合分数与温度、密度和平衡产物质量分数等的关系。目前,对金属Li 的相关反应机理鲜有研究,化学反应动力学数据更是缺乏。
Li/SF6燃烧反应实验主要用于观测射流尺寸、温度变化以及产物分布。Avery 和Facth[15]通过实验获得了反应区尺寸、扩展速率及射流中心线处的温度分布,发现高金属密度的射流导致蒸汽相的出现,进而发生凝结和扩散;Parnell 等[16]利用X 光照相技术观测了反应区尺寸、稳定性及扩展速率,结果表明射流长度与熔融锂浓度有关;Hsu 等[17]实验观测了点火及燃烧过程,将其分为点火、火焰传播、稳定燃烧和熄火4 个阶段;郑邯勇等[18]利用X 光高速动态成像技术观测了Li/SF6浸没喷射反应过程,测量了SF6在液态锂中的浸没喷射长度和宽度,建立了描述SF6在液态锂中浸没喷射长度的半经验关系式。李维维等[19]开展了Li/SF6的小功率锅炉反应器快速启动试验,研究了点火后氧化剂和冷却水进入时序、流量匹配和启动剂产物等对化学反应和换热的影响。由于燃烧实验成本大、测量困难,而单独金属颗粒的燃烧无法体现反应组分间相互作用,Li/SF6浸没燃烧实验难度极大。
数值仿真可以预估实验难测量参数,节省研究费用,缩短研究周期,指导反应器结构设计及参数优化,有利于提高系统可靠性和经济性,因此广泛应用于Li/SF6燃烧反应研究。Chan 等[20]应用均质流(homogeneous flow,LHF)模型仿真发现反应距离很短,但射流深度大,同时研究了Li 局部低温对流动结构的影响;Chan 等[21]研究发现,与LHF 模型相比,多流体模型仿真得到的射流深度与实验数据更为接近;Dahikar 等[22]研究了喷嘴直径及布置方式、射流气体速度、熔融Li 温度对喷射尺寸的影响,发现喷嘴水平放置对反应更有利,可以得到稳定的射流长度;Chen 等[23]建立了灯芯式燃烧的数学模型,阐明雷诺数是影响强制对流燃烧的重要参数,气流速度增加使得总燃烧速率增加;Gulawani 等[24]研究发现传质系数与湍动能及湍流扩散率有关,基于多流体模型的浸没喷射流型、射流尺寸及温度等的结果与实验数据更为相符。
尽管数值仿真在重现实验过程和数据方面取得了进展,现有针对低功率密度Wick 燃烧和高功率密度浸没燃烧仿真的燃烧反应模型误差仍较大,这一方面是因为未考虑辐射换热,导致温度场的准确性降低,另一方面是缺乏化学反应机理认识,Li/SF6反应活化能、反应热、反应速率以及扩散系数等热/动力学参数的设置缺乏理论指导。目前对于Li/SF6反应的中间步骤认识不清,只能采用基元反应代替反应过程,计算误差较大。因此通过分子仿真的手段来揭示金属燃料反应机理是非常有必要的。基于反应力场(reactive force field,ReaxFF)的分子动力学仿真完全由体系势能推动,计算过程中无须预设反应路径,非常适用于分析反应体系的微观机理。
ReaxFF[25-26]能够在原子水平上描述反应物和产物在不同条件下的结构变化和化学反应等过程,通过分析反应过程中组分的变化可以得到反应路径及中间产物等重要信息。为了揭示Li/SF6燃烧的分布反应机理,文中结合ReaxFF 反应分子动力学仿真和第一性原理计算方法研究了热源启动过程中Li 和SF6的微观反应过程,分析了主要反应物和产物组分的动态演化特性,获得其主要反应路径及反应热,为构建或验证Li/SF6燃烧反应动力学模型提供机理认识和理论基础。
1 分子动力学仿真模型及方法
1.1 仿真模型
首先,采用Materials Studio 软件构建Li 和SF6单分子的几何模型,并进行结构优化;其次,采用Amorphous Cell 模块构建包含多个Li 和SF6分子的周期性盒子(初始尺寸为7×7×7 nm3)作为仿真模型,一定数目的Li 和SF6分子在盒子内随机分布;再次,根据初始温度条件合理设定反应物的初始密度以避免原子重叠,通过改变盒子中Li/SF6分子数目及比例来控制反应物组分的质量比;最后,利用Forcite 模块在等温等压系综(NPT)和正则系综(NVT)下对特定温度(500 K)和压力(0.1 MPa)条件下的反应体系进行50 ps 的非反应动力学平衡过程仿真,从而完成分子动力学仿真模型(见图1,图中红色为S 原子,蓝色为F 原子;绿色为Li 原子)的构建。

图1 Li/SF6 反应的分子动力学仿真模型Fig.1 Molecular dynamics simulation model of Li/SF6 reaction
1.2 仿真方法
上述分子动力学仿真模型随后采用LAMMPS软件中的ReaxFF 程序包开展分子动力学仿真,所用力场参数为Islam 等[27]拟合开发的C/O/H/F/Li/S体系力场参数。该计算力场中C/H/O/S 和Li 参数,以及F 参数是基于已有文献中的力场参数,并经量子化学计算进行了额外的力场训练,能够准确描述键解离、Li-F 相互作用,以及其他分子与Li 和F 的结合。计算过程中采用周期性边界条件和微正则系综(NVE),时间步长为0.25 fs,采用Berendsen 方式控制体系温度,温度阻尼系数设置为10 fs。仿真工况如表1 所示。每个工况进行3 次平行仿真以获得更好的统计学结果。仿真完成后,采用键级截断的方法判断原子成键及反应产物,统计分析原子轨迹、关键产物及化学键等相关信息,追踪特征产物组分随反应过程的动态演化规律,进而分析总反应中各个分反应的内在联系及其主次规律。
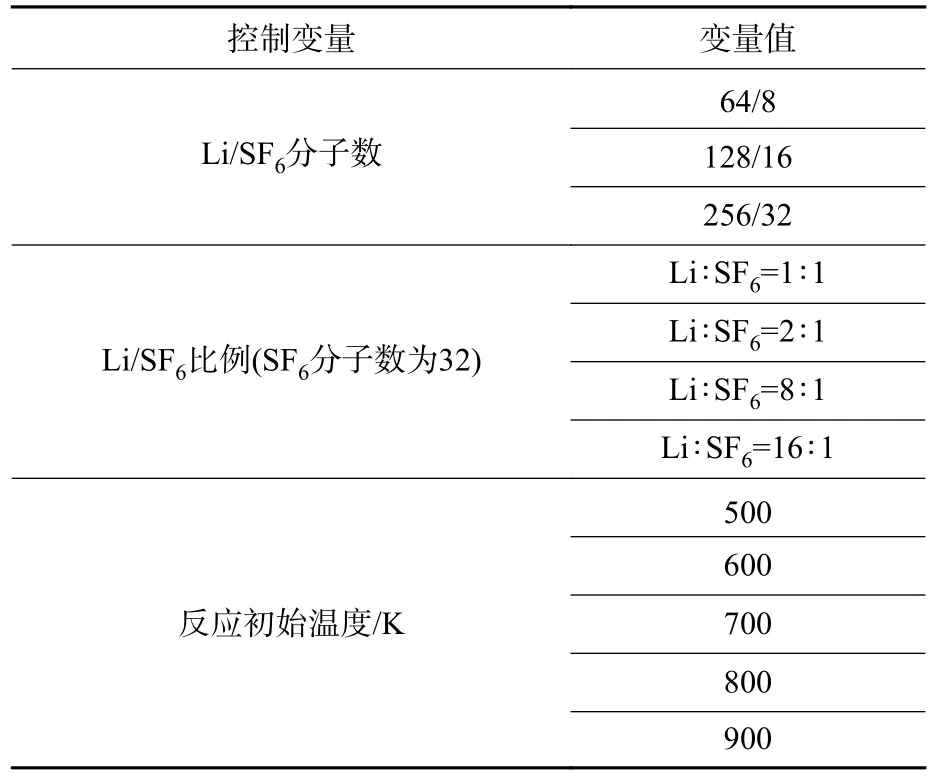
表1 Li/SF6 反应仿真工况Table 1 Simulation conditions of Li/SF6 reaction
1.3 第一性原理计算
从量子力学理论出发的第一性原理计算方法,根据原子核和电子相互作用的原理及其基本运动规律,近似处理后直接求解薛定谔方程,能够得到物质的基态性质。因此文中采用第一性原理计算Li/SF6反应过程中各物质的基态性质。
采用Material Studio 软件中的DMol3 模块[28]对反应过程的热力学性质进行分析计算。通过得到体系的总电子和离子能量Etotal以及焓值的修正值Htotal之后,计算可得反应物/产物总的焓值H=Etotal+Htotal。体系的总电子和离子能量Etotal包含于体系内能U中,内能由电子、振动、平移和旋转分量组成,即
在反应焓值计算过程中,首先建立单独的分子模型并采用DMol3 模块对分子结构进行几何优化,得到分子的基态结构;之后使用统计力学获得简正模的振动频率,并对分子进行振动分析计算,即可获得式(1)中的各项数值;利用DMol3 中的analysis 功能计算得到重要的热力学性质,从中读取修正焓值Htotal与温度之间的函数关系,得到在特定温度T下的焓值修正值Htotal;计算得到各个分子的焓值之后,根据反应热的计算公式求得总的反应热。
反应热∆H可以利用反应物焓值Hreactatns与生成物焓值Hproducts之差进行计算,即
2 仿真结果与分析
2.1 主要反应路径分析
为了解Li/SF6反应的主要路径,文中设置了多组不同SF6和Li 分子数目的工况进行对比分析,在当前仿真盒子尺寸条件下,SF6和Li 分子数分别为8 和64 时便能获得相对稳定的结果。图2(a)展示了特定工况(反应物分子数SF6和Li 比例为8∶64,初始温度为500 K)下仿真系统的最终构象,可以看出反应生成了固体产物LiF 和Li2S。图2(b)给出了该工况下主要反应物和产物组分随仿真时间的变化情况。从反应过程中各个组分的分子数变化及反应分子动态视图可知,反应的起始过程为SF6分子S-F 键的断裂,LiF 是反应初始时刻的主要产物;随着反应的进行,多余的Li 反应形成Li2或与S 成键形成Li2S;反应后期,LiF 分子间发生反应结合生成Li2F2或Li3F3。由此可以给出Li/SF6反应的主要反应路径(见图2(b)): SF6→S+6F、Li+F→LiF、2Li→Li2、Li2+S→Li2S、2LiF→Li2F2。
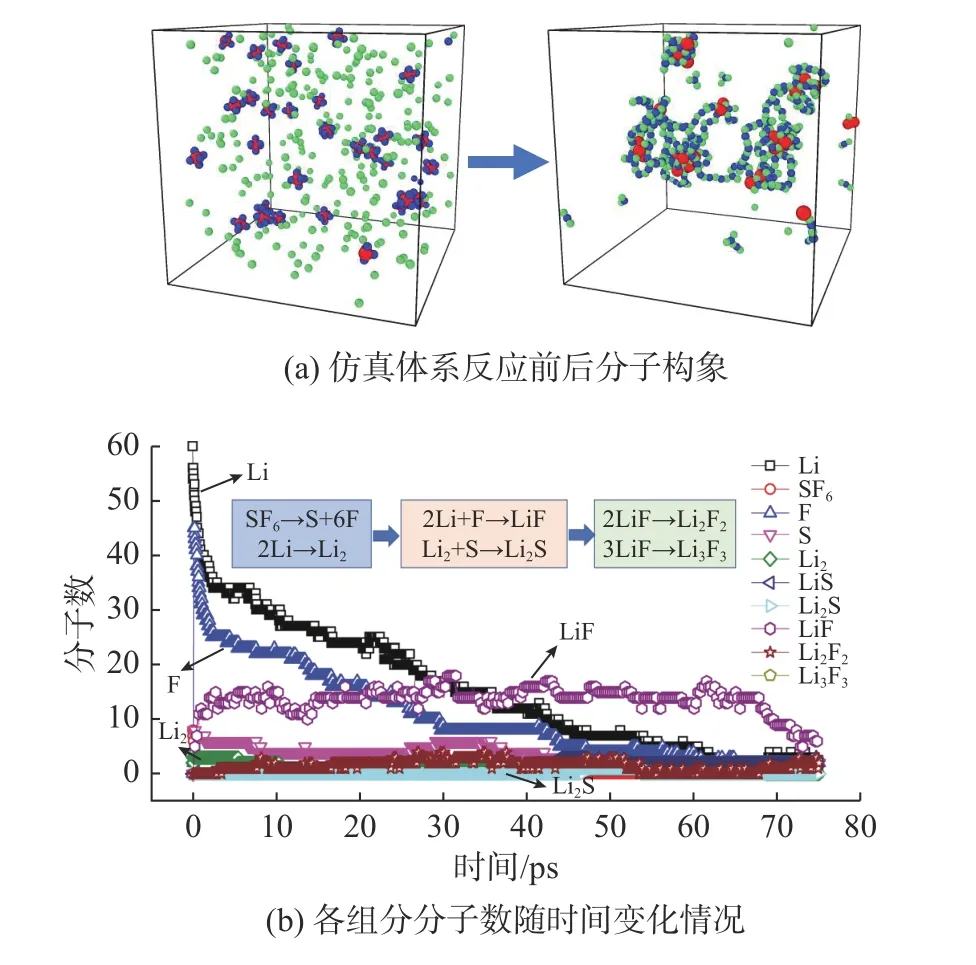
图2 Li/SF6 反应前后系统构象及体系中组分分子数随时间变化情况Fig.2 Conformation of Li/SF6 system before and after reaction and variation of molecule number with time
由于温度是影响反应的关键因素,因此文中首先研究了不同初始温度对反应过程的影响。图3展示了反应物分子数SF6和Li 比例为8∶64 时,不同初始温度下Li 和LiF 分子数随时间的变化情况。由图可知,初始温度对反应初始阶段影响较小,这是因为反应初始阶段主要为SF6分解,SF6分解速率在当前仿真温度(500~900 K)相差不大。但是随着初始温度增加,Li 消耗速率增加,同时反应接近平衡时的Li 剩余比例也较低,表明更多的Li 与其他原子发生反应,总反应也进行得更为彻底。这一点从图中LiF 的分子数变化也可以推测得到。随着反应初始温度增加,更多的Li 与F 反应生成LiF,因此初始温度越高,反应趋近平衡时的LiF 分子数越多。由于文中重点关注反应的初始阶段,温度对反应的影响放在后续研究中,这里不再赘述。

图3 不同初始温度下Li 和LiF 分子数随仿真时间变化情况Fig.3 Variation of Li and LiF molecule number with simulation time under different initial temperatures
文中同时研究了反应物浓度对Li/SF6反应过程的影响。图4 展示了在相同仿真盒子尺寸、反应物比值(SF6∶Li=1∶8)和初始温度(500 K)条件下Li、F、LiF 这3 种主要组分随时间的变化情况。由图可知,随着反应物浓度增加,反应进行得越快,反应接近或达到平衡所需的时间越短,这是由于随着反应物浓度增加,分子间距离缩短,反应物分子间的碰撞概率增加,使得反应加快。产物LiF 的变化情况则较为复杂,不仅受到前序反应(SF6分解)的影响,还与LiF 的后续反应有关: 反应初期,LiF 分子数随反应物浓度的增加而增大;随着反应的不断进行,LiF 分子数越大,碰撞概率越大,不断结合反应生成Li2F2,因此反应物浓度越高的体系中LiF 分子数反而越小。

图4 不同反应物浓度下Li、F 和LiF 分子数比例随时间变化情况Fig.4 Variation of proportion of Li,F,and LiF molecule number with time under different reactant concentrations
文中也分析了不同仿真盒子尺寸和温度(500 K)条件下,维持SF6分子数不变,通过改变反应物Li 的分子数来分析反应物比例对反应过程的影响。图5 展示了反应物Li 和主要产物LiF 数目随反应时间的变化情况。由图5 可知,随着Li 分子数增加,其与S、F 原子发生碰撞概率增加,反应物Li 消耗速率增加;反应产物LiF 在反应初始阶段随Li 分子数增加而增加,反应后期多余的Li 与LiF 继续反应生成Li2F2和Li3F3,反而导致LiF 分子数减少,这也从侧面证明了上述主要反应路径的合理性。

图5 不同反应物比例下Li 和LiF 分子数占比随时间变化情况Fig.5 Variation of proportion of Li and LiF molecule number with time under different reactant proportions
2.2 反应速率计算
通过分析反应物Li 的浓度变化来获得总反应的反应速率。在容积不变的反应容器里,反应速率v(mol·L-1·s-1)定义为单位体积内反应进度(ε=Δn/ν)对时间的变化率,即
式中:V为体积;v 为化学计量数;n为物质的量;c为物质的量浓度。
图6 给出了不同反应条件下Li/SF6总反应速率随时间的变化情况。由图可知,反应速率与反应物浓度或分子数呈正相关关系,在相同反应容积下,反应物浓度越大,反应物分子间距离越小,碰撞概率越高,因此反应速率越大;Li 在很短时间内(≈10 ps)被反应消耗掉,反应速率随着Li 的消耗迅速减小后趋于稳定;随着反应物比例(或Li 分子数)增大,反应速率几乎呈同比例线性增加,因为随着Li 分子数增加,Li 与其他原子碰撞发生反应的概率也增加;反应初始温度对总反应速率的影响较小,但从LiF 分子数变化可以推断初始温度对分步反应的反应速率是有影响的。
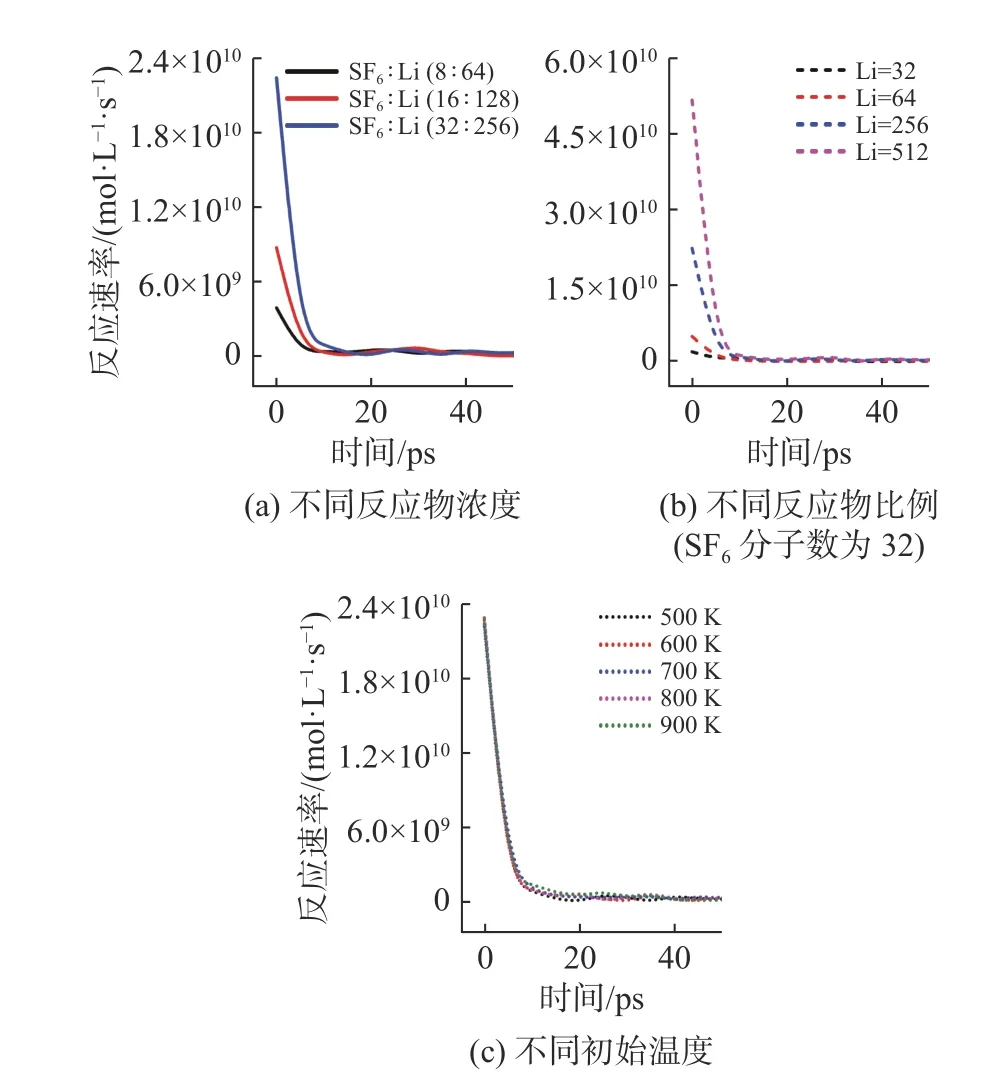
图6 不同反应条件下反应速率随时间变化情况Fig.6 Variation of reaction rate with time under different reaction conditions
2.3 反应热计算
根据上述确定的分反应,采用第一性原理计算获得每个分反应对应的放热量。反应过程中各分子的能量折线图和分子示意图如图7 所示。

图7 反应过程中反应物的能量折线图及分子示意图Fig.7 Energy line diagram and molecular diagram of reactants in reaction process
上述分析得到的反应SF6→S+6F、Li+F→LiF、2Li→Li2、Li2+S→Li2S 为总反应的分反应,而2LiF→Li2F2为总反应产物的双分子结合反应,根据盖斯定律[29]可得总反应的反应热计算公式为
反应2LiF→Li2F2的反应热计算公式为
因此整个化学反应过程的反应热为上述2 部分反应热的总和。需要注意的是生成1 个Li2F2分子需要2 个LiF 分子,而总反应中共生成6 个LiF 分子,因此需要调整反应热的系数,即
由上式可知,为了计算反应的生成热,需要计算得到各个反应物/产物的焓值。采用第一性原理计算获得了各个反应物/产物分子的焓值,如表2所示。
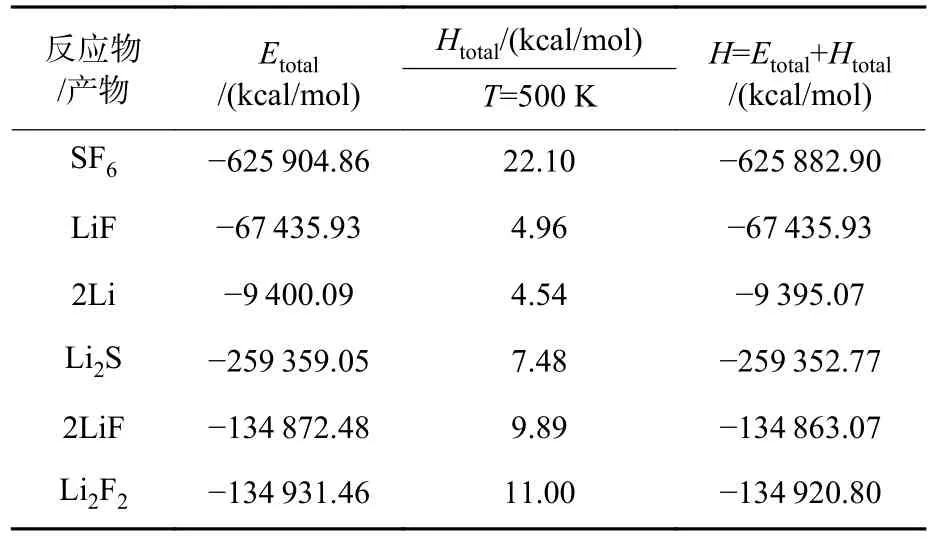
表2 Li/SF6 反应过程中反应物/产物焓值计算结果Table 2 Calculation results of reactant/product enthalpy value during Li/SF6 reaction
根据上表计算所得焓值,可通过以下公式计算得到Li/SF6反应的反应热:
根据反应物/产物标准焓值计算得到的反应热为-2 318 kJ/mol,文献中实验测得的反应热为-2 621 kJ/mol[18],均与文中计算结果接近。计算得到的反应热与理论值及实验值之间的误差主要来源于仿真温度、压力等条件与实际反应条件的差异,以及仿真采用的反应力场与实际反应过程的吻合程度。表明通过分子仿真获得主要反应路径,然后再基于第一性原理计算得到复杂反应过程的反应热具备一定的可靠性。
3 结论
文章采用ReaxFF 反应分子动力学仿真方法研究了Li 和SF6微观反应过程,获得产物分布和动态演化特性,以及其主要反应路径,然后结合第一性原理计算得到了主要反应路径的反应热,主要结论有以下几个方面。
1) Li/SF6反应的起始过程为SF6分子S-F 键的断裂,LiF 是反应初始时刻的主要产物;随着反应的进行,多余的Li 形成Li2与S 成键形成Li2S;反应后期,LiF 分子间结合反应形成Li2F2。由此判断Li/SF6反应主要路径为: SF6→ S+6F、Li+F →LiF、2Li→Li2、Li2+S→Li2S、2LiF→Li2F2。
2) 获得了反应物浓度、比例以及初始温度对反应速率的影响规律: 在相同容积条件下,反应速率大小与反应物浓度呈正相关,浓度越大,反应物分子间距离越小,发生碰撞概率越高,因此反应速率越大;随着反应物比例增大,反应速率几乎呈同比例线性增加,因为Li 与其他原子碰撞概率增加;初始温度对总反应速率的影响较小,但对分步反应的反应速率是有影响的。
3) 采用第一性原理计算获得Li/SF6反应主要路径或分步反应对应的放热量,进而获得Li/SF6反应的反应热为-2 216.7 kJ/mol,表明通过分子仿真获得主要反应路径,然后结合第一性原理计算得到反应热是可行的,可用于复杂燃烧反应机理揭示及反应热计算。
总之,文中建立了能够描述Li 和SF6微观反应过程的分子仿真方法,结合第一性原理计算实现了Li 和SF6反应的主要路径判断及反应热计算,分析了反应物浓度、反应物比例及初始温度对总反应速率的影响规律,揭示了Li 和SF6燃烧反应的微观机制。研究结果为揭示复杂燃烧反应的微观机理和反应热计算提供了有效手段,获得了Li/SF6燃烧反应的反应热、反应速率等关键动力学及热力学参数,为构建或验证Li/SF6燃烧反应动力学模型提供了机理认识。
但是文中一方面尚未揭示反应温度对反应效率以及反应路径的影响规律和机制,后续研究需着重针对此方面展开。另一方面,由于Li/SF6燃烧反应过程非常复杂,受温度差异、时空尺度差异以及力场极化等因素的影响,当前基于ReaxFF 力场的分子动力学仿真模型还难以直接服务于燃烧反应微观模型的建立,笔者正通过结合伞状采样方法开展加速分子动力学仿真,尝试建立介尺度燃烧反应分子模型;同时,鉴于微观反应与宏观多相流动在时间和空间尺度上的巨大差异,当前微观尺度的仿真研究还难以考虑宏观尺度流体动力学的影响,亟需发展兼顾微观反应和宏观流动的介观尺度或多尺度仿真技术,结合统计物理方法,建立一个可被移植到宏观仿真的微观模型与工具,在微观反应机理与宏观燃烧过程间搭建起沟通的桥梁,从而更好地指导燃烧反应器的优化设计与应用。
