含1,2,4-噁二唑噻唑类化合物的设计合成及生物活性
2023-12-06张如松姜硕朱宝玉光明甲王明慧许良忠孙鉴昕
张如松,姜硕,朱宝玉,光明甲,王明慧,许良忠,孙鉴昕
(青岛科技大学 化学与分子工程学院,山东 青岛 266042)
在新农药的创制中,氮杂环类化合物[1-2]占到了所有新化合物的80%,其中噻唑以及噁二唑都占了很大的比重,噻唑及噁二唑类化合物在农药方面可以用作杀菌剂、杀螨剂、杀虫剂、除草剂,具有广谱的生物活性。
过去已经报道了很多种商品化的噻唑类化合物和噁二唑类化合物,如噻虫嗪、噁虫酮、噁草酮、噻菌灵等,目前对此类化合物的研究热度依然不减,Sun等[3]人设计合成了一系列含1,3,4-噁二唑杂环结构的化合物,该系列所有化合物在质量浓度为500 mg/L时,对黏虫的致死率为100%,部分化合物在100 mg/L时,仍能保持100%的致死率;部分化合物在500 mg/L时对蚜虫的致死率为100%,少部分化合物在100 mg/L时对蚜虫的致死率能在70%以上;Wang等[4]设计合成了一系列新型1,2,4-噁二唑类化合物,其中一个化合物在质量浓度为0.39125 mg/L时,对大豆锈病的抑制率仍能达到90%;Ma等[5]设计合成了一系列芳基噻唑胺类衍生物,部分化合物具有广谱的抑菌活性,并且抑制率优于商品化试剂噁菌灵和百菌清;Zhou等[6]设计合成了一系列4-芳基-3-羟基异噻唑衍生物,部分目标化合物对斜纹叶蛾幼虫具有一定的杀虫活性。
为了解决目前农药抗性的问题,以噻虫嗪及噁虫酮为先导化合物,通过活性亚结构拼接法[7],连接一系列具备生物活性的基团,合成出了一系列含1,2,4-噁二唑噻唑类化合物,以期获得新的先导化合物,设计路线见图1。
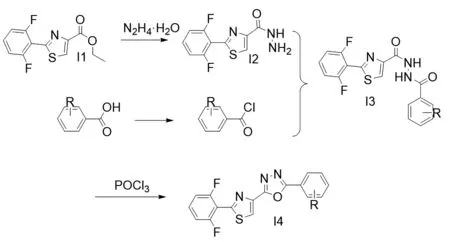
图1 目标化合物的设计路线
1 实验部分
1.1 主要仪器和试剂
X-4显微熔点仪,上海精密科学仪器有限公司;ZF-2紫外分析仪,上海市安亭电子仪器厂;BRUKER AVANCE500Mhz核磁共振仪,德国-瑞士布鲁克光谱仪器公司;HRMS高分辨质谱仪,赛默飞世尔科技有限公司;RE-52AA旋转蒸发仪,青岛蓝特恩科教仪器设备有限公司;SHZ-D(III)循环水式真空泵,青岛蓝特恩科教仪器设备有限公司;YP1002N电子天平,上海驰唐仪器设备有限公司;DL SB-5L/20低温冷却循环泵。青岛蓝特恩科教仪器设备有限公司;CT-2恒温加热磁力搅拌器,郑州豫华仪器制造有限公司;DHG-9070A电热鼓风干燥箱,青岛蓝特恩科教仪器设备有限公司;SB-1100机械搅拌器,青岛蓝特恩科教仪器设备有限公司;LRH-150F智能光照培养箱,上海一恒科学仪器有限公司。
2,6-二氟苯甲酸、丙酮酸乙酯、N-溴代丁二酰亚胺、对叔丁基苯甲酸、2-氯烟酸、3,5-双三氟甲基苯甲酸、2-甲基-3-硝基苯甲酸、2,3,4,5,6-五氟苯甲酸,分析纯,上海毕得医药科技有限公司;甲苯、乙腈、乙醇、水合肼、三乙胺、乙酸乙酯、石油醚,分析纯,烟台双双化工。
1.2 合成路线及方法
目标化合物的合成路线见图2。
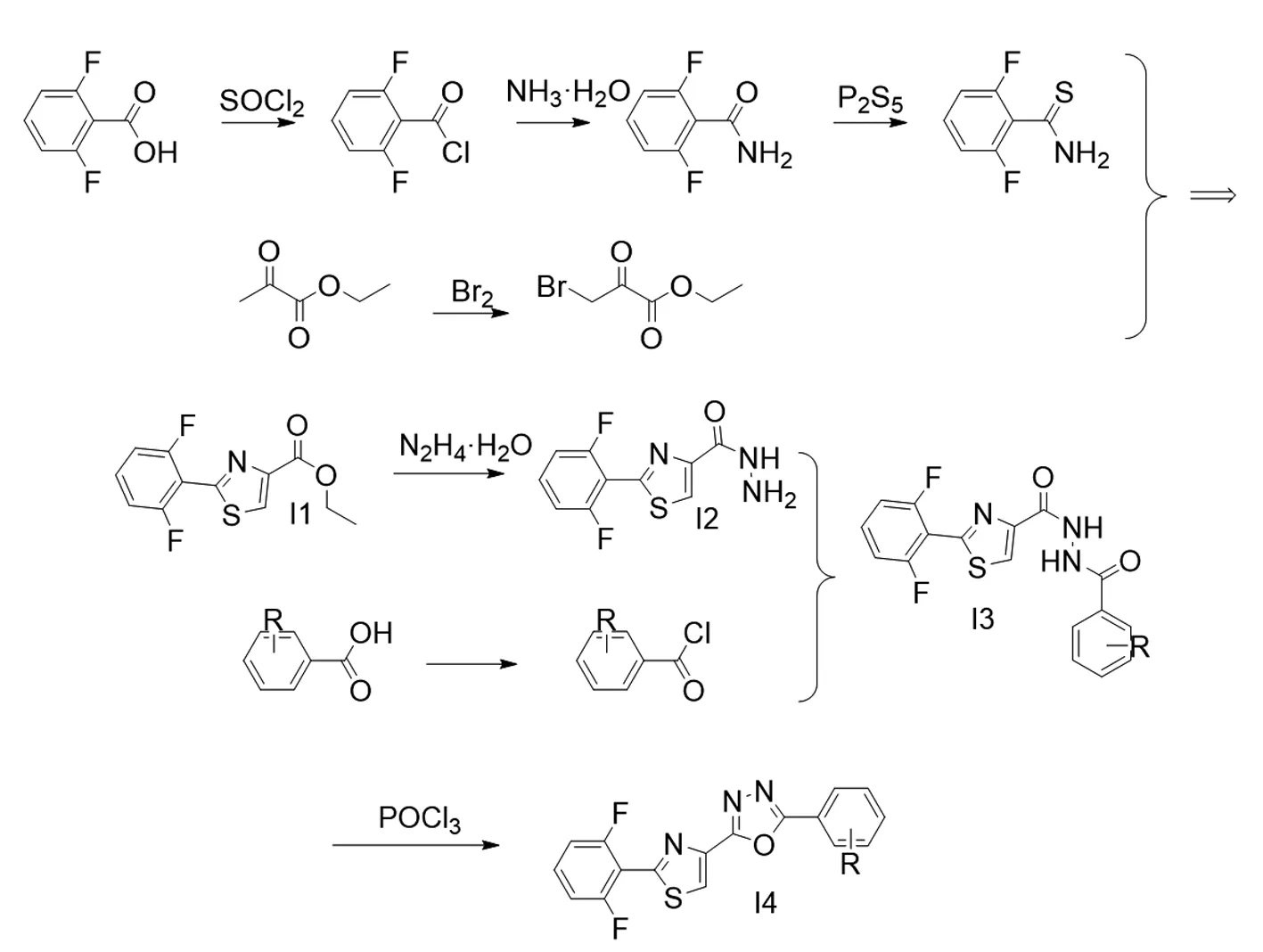
Ra=4-tbu;Rb=2-Cl py;Rc=3,5-CF3;Rd=2-Me,3-NO2;Re=2,3,4,5,6-F
1.2.1 中间体2,6-二氟苯并硫代酰胺的合成
在250 mL的三口烧瓶中,首先加入15.8 g(0.1 mol)2,6-二氟苯甲酸,然后加入80 mL甲苯作为反应溶剂,再加入13.6 g(0.115 mol)氯化亚砜,添加回流冷凝管,搅拌,加热反应至回流,在回流状态下,反应4 h,TLC监测反应进度,待反应完全后,减压蒸馏除去多余溶剂及氯化亚砜,得到中间体2,6-二氟苯甲酰氯17.25 g,为淡黄色油状液体,收率98%,不经处理用于下一步反应;另取一250 mL三口烧瓶,首先加入100 mL氨水,然后将反应瓶置于冷肼中,控制冷肼温度为0 ℃,搅拌,在1 h内缓慢向反应液中滴加17.6 g(0.1 mol)2,6-二氟苯甲酰氯的乙腈溶液(质量分数40%),滴加完毕后,将反应瓶转移至室温条件,继续反应2 h,TLC监测反应进度,待反应完全后,将反应液抽滤,取滤饼,烘干得白色固体2,6-二氟苯甲酰胺15.2 g,收率96.8%,不经处理用于下一步反应;再取一250 mL三口烧瓶,首先加入15.7 g(0.1 mol)2,6-二氟苯甲酰胺,再加入60 mL无水乙醇作为反应溶剂,最后加入8.88 g五硫化二磷(0.04 mol),搅拌,缓慢升高反应温度至50 ℃,保持温度反应12 h,TLC监测反应进度,待反应完全后,降温,减压蒸馏除去多余乙醇,然后加入60 mL水,以60 mL乙酸乙酯分3次(3×20 mL)分批萃取,合并有机相,用无水硫酸钠干燥,减压蒸馏得红色固体中间体 2,6-二氟苯并硫代酰胺14.7 g,收率85%,不经提纯,直接用于下一步反应。
1.2.2 中间体溴代丙酮酸乙酯的合成
取一个250 mL三口烧瓶,首先加入11.6 g(0.1 mol)丙酮酸乙酯,然后加入50 mL二氯甲烷作为反应溶剂,搅拌,在常温条件下缓慢滴加16 g(0.1 mol)溴素,每滴入一滴,待其颜色褪去后,再继续滴入下一滴,滴加完毕后,在常温条件下继续反应2 h,TLC监测反应进程,待反应完全后,向反应体系中加入20 mL饱和亚硫酸钠水溶液,分层取有机相,以60 mL(3×20 mL)饱和食盐水洗涤有机相,用无水硫酸钠干燥,减压蒸馏除去多余溶剂,得溴代丙酮酸乙酯18.5 g,淡黄色液体,收率95.4 %。
1.2.3 中间体2-(2,6-二氟苯基)噻唑-4-甲酸乙酯的合成
取一个250 mL三口烧瓶,首先加入19.4 g(0.1 mol)溴代丙酮酸乙酯,然后加入50 mL无水乙醇作为反应溶剂,再加入17.3 g(0.1 mol)2,6-二氟苯并硫代酰胺,搅拌,缓慢将温度升至回流,在回流条件下继续反应12 h,TLC监测反应进度,待反应完全后,降温,减压蒸馏除去大部分乙醇,放置0.5 h,抽滤析出的固体,用无水乙醇(3×5 mL)洗涤固体,收集滤液留作下次套用,滤饼烘干后得2-(2,6-二氟苯基)噻唑-4-甲酸乙酯16.7 g,白色固体,收率62%(套用后收率在85%)。
1.2.4 中间体2-(2,6-二氟苯基)噻唑-4-甲酰肼的合成
取一个250 mL三口烧瓶,首先加入26.9 g(0.1 mol)中间体 2-(2,6-二氟苯基)噻唑-4-羧酸乙酯,然后加入50 mL乙醇作为反应溶剂,再加入25 g(0.4 mol)水合肼(含量80%),搅拌,缓慢升温至回流,在回流状态下继续反应4 h,TLC监测反应进度,待反应完全后,降温,待温度降至室温时,向三口烧瓶中加入100 mL水,此时析出大量固体,抽滤,并用30 mL(3×10 mL)水洗涤滤饼,收集滤饼并烘干,得24.7 g中间体2-(2,6-二氟苯基)噻唑-4-甲酰肼,淡黄色固体,收率96.9%。
1.2.5 中间体对叔丁基苯甲酰氯的合成
取一个250 mL的三口烧瓶,首先加入17.8 g(0.1 mol)对叔丁基苯甲酸,然后加入50 mL的二氯甲烷作为溶剂,搅拌,在室温条件下1 h内缓慢滴加15.1 g(0.12 mol)草酰氯,滴加完毕后,继续在室温条件下反应4 h,TLC监测反应进程,待反应完全后,减压蒸馏除去多余溶剂二氯甲烷及过量的草酰氯,得对叔丁基苯甲酰氯18.4 g,黄色液体,收率94%。
其余中间体2-氯烟酰氯、3,5-双三氟甲基苯甲酰氯、2-甲基-3-硝基苯甲酰氯、2,3,4,5,6-五氟苯甲酰氯的合成均使用上述方法合成。
1.2.6 中间体N-(4-(叔丁基)苯甲酰基)-2-(2,6-二氟苯基)噻唑-4-甲酰肼的合成
取一个250 mL的三口烧瓶,首先加入5.1 g(0.02 mol)2-(2,6-二氟苯基)噻唑-4-甲酰肼,然后加入30 mL乙腈作为反应溶剂,再加入2.2 g(0. 22 mol)三乙胺作为缚酸剂,搅拌,室温滴加4.1 g(0.21 mol)对叔丁基苯甲酰氯,滴加完毕后升温至40 ℃反应4 h,TLC监测反应进度,反应完全后,降温至室温,向三口烧瓶中加入30 mL水,搅拌0.5 h,抽滤析出的固体,用15 mL(3×5 mL)水洗涤滤饼,得中间体N-(4-(叔丁基)苯甲酰基)-2-(2,6-二氟苯基)噻唑-4-甲酰肼7.85 g,白色固体,收率92.4%。
其余中间体N-(3-氯吡啶酰基)-2-(2,6-二氟苯基)噻唑-4-甲酰肼、N-(3,5-双(三氟甲基)苯甲酰基)-2-(2,6-二氟苯基)噻唑-4-甲酰肼、N-(2-甲基-3-硝基苯甲酰基)-2-(2,6-二氟苯基) 噻唑-4-甲酰肼、N-(全氟苯甲酰基)-2-(2,6-二氟苯基) 噻唑-4-甲酰肼的合成均使用上述方法合成。
1.2.7 目标化合物2-(4-(叔丁基)苯基)-5-(2-(2,6-二氟苯基)噻唑-4-基)-1,3,4-噁二唑的合成
取一个250 mL的三口烧瓶,首先加入4.15 g(0.01 mol)中间体N-(4-(叔丁基)苯甲酰基)-2-(2,6-二氟苯基)噻唑-4-碳酰肼,然后加入15 mL甲苯作为反应溶剂,再加入6.08 g(0.04 mol)三氯氧磷,搅拌,逐渐升温至回流,在回流条件下继续反应6 h,TLC监测反应进程,待反应结束后,降温至室温,向三口烧瓶中加入40 mL水,用45 mL(3×15 mL)乙酸乙酯分批萃取,取有机相,加无水硫酸钠干燥,减压蒸馏得到粗品,经柱层析(V(乙酸乙酯)∶V(石油醚)=1∶15) 分离,得到目标化合物2-(4-(叔丁基)苯基)-5-(2-(2,6-二氟苯基)噻唑-4-基)-1,3,4-噁二唑,为白色固体。
其余目标化合物2-(3-氯吡啶-2-基)-5-(2-(2,6-二氟苯基)噻唑-4-基)-1,3,4-噁二唑、2-(3,5-双(三氟甲基)苯基)-5-(2-(2,6-二氟苯基)噻唑-4-基)-1,3,4-噁二唑、2-(2-(2,6-二氟苯基)噻唑-4-基)-5-(2-甲基-3-硝基苯基)-1,3,4-噁二唑、-(2-(2,6-二氟苯基)噻唑-4-基)-5-(全氟苯基)-1,3,4-噁二唑的合成均使用上述方法。
化合物的结构表征数据如下:
2-(4-(叔丁基)苯基)-5-(2-(2,6-二氟苯基)噻唑-4-基)-1,3,4-噁二唑(I4a):白色固体 m.p.129.8~132.7 ℃,1H NMR (500 MHz,DMSO-d6) δ 8.95 (s,1H),8.03 (d,J=8.5 Hz,2H),7.74~7.67 (m,1H),7.65 (d,J=8.3 Hz,2H),7.38 (t,J=8.7 Hz,2H),1.33 (s,9H).13C NMR (101 MHz,DMSO-d6) δ 164.34,161.19,160.19,158.67,156.74,155.58,140.09,133.48,127.05,126.69,120.87,113.25,113.00,110.66,35.28,31.22. ESI HRMS calcd for C21H17F2N3OS ([M+H]+) 398.113 9,found398.113 2。
2-(3-氯吡啶-2-基)-5-(2-(2,6-二氟苯基)噻唑-4-基)-1,3,4-噁二唑(I4b):白色固体,收率86 %,m.p.174~176 ℃1H NMR (500 MHz,Chloroform-d) δ 8.63 (d,J=4.4 Hz,1H),8.53~8.47 (m,2H),7.48 (dq,J=8.9,4.7,4.1 Hz,2H),7.13 (t,J=8.7 Hz,2H).13C NMR (126 MHz,DMSO-d6) δ 161.01,160.87,160.71,158.80,156.96,151.41,148.98,139.63,139.52,131.48,124.16,121.92,119.84,111.89,111.70,110.39. ESI HRMS calcd for C16H7ClF2N4OS ([M+H]+) 377.007 5,found377.007 1。
2-(3,5-双(三氟甲基)苯基)-5-(2-(2,6-二氟苯基)噻唑-4-基)-1,3,4-噁二唑(I4c):白色固体,收率91 %,m.p.116~118 ℃1H NMR (500 MHz,DMSO-d6) δ 9.08 (s,1H),8.62 (s,2H),8.42 (s,1H),7.73~7.63 (m,1H),7.36 (t,J=8.7 Hz,2H).13C NMR (126 MHz,DMSO-d6) δ 161.71,160.49,160.36,158.38,156.43,139.12,132.99,131.29,130.33,127.25,127.07,125.77,125.33,121.62,113.70,112.67,112.49,110.02,99.51. ESI HRMS calcd for C19H7F8N3OS ([M+H]+) 478.026 0,found478.025 3。
2-(2-(2,6-二氟苯基)噻唑-4-基)-5-(2-甲基-3-硝基苯基)-1,3,4-噁二唑(I4d):白色固体,收率89 %,m.p.144.6~146.8 ℃1H NMR (500 MHz,Chloroform-d) δ 8.49 (s,1H),8.25 (d,J=7.9 Hz,1H),7.91 (d,J=8.0 Hz,1H),7.58~7.43 (m,3H),7.10 (t,J=8.7 Hz,2H),2.85 (s,3H).13C NMR (101 MHz,Chloroform-d) δ 168.83,163.29,160.61,159.07,157.43,152.23,140.16,134.26,133.30,132.71,132.05,126.96,126.52,124.60,112.48,112.23,110.86,16.99. ESI HRMS calcd for C18H10F2N4O3S ([M+H]+) 401.052 0,found401.051 2。
2-(2-(2,6-二氟苯基)噻唑-4-基)-5-(全氟苯基)-1,3,4-噁二唑(I4e):白色固体,收率85 %,m.p.196.8~199 ℃1H NMR (500 MHz,Chloroform-d) δ 9.88 (s,1H),8.86 (ddd,J=14.7,8.4,6.2 Hz,1H),8.49 (t,J=8.5 Hz,2H).13C NMR (101 MHz,Chloroform-d) δ 161.63,161.58,161.51,159.08,159.03,157.59,139.73,132.17,132.06,131.95,125.10,125.07,125.04,112.46,112.44,112.24,112.21.ESI HRMS calcd for C17H4F7N3OS ([M+H]+) 432.004 2,found432.002 6。
1.3 生物活性测试
1.3.1 对二斑叶螨卵室内活性测试
对所合成的目标化合物参照 NY/T 1154.5—2006《农药室内生物测定试验准则杀虫剂》第 5 部分:杀卵活性试验浸渍法进行了生物活性测试。具体方法如下:按照试验设计将药剂配制成为系列浓度的稀释液,以 0.1%吐温-80 水为对照;取长势相同的辣椒苗盆栽,留中心展平的 4 片叶,在叶柄处涂抹凡士林,防治成螨逃逸;每片叶接 20 头成螨,在养虫室放置 24 h;第二天将成螨挑下,用体视镜查清每片叶上的卵数并记录,将叶片摘下,浸药并晾干后置于盛有琼脂的培养皿中,叶背面朝上,置于密封袋中保湿并培养。放入培养箱中进行培养。每个浓度重复2次。置于(26±1) ℃、相对湿度 70%~80%、光照 16 h∶8 h(L∶D)条件下正常饲养,药后7 d记录未孵化卵数,计算孵化抑制率和校正孵化抑制率。二斑叶螨卵数据处理方法采用 Excel 数据处理软件对各处理的孵化率和孵化抑制率进行计算和分析。
孵化率=孵化数/处理总卵数×100%
(1)
孵化抑制率=(对照孵化率-处理孵化率)/对照孵化率×100%
(2)
2 结果与讨论
2.1 对二斑叶螨卵的室内生物测试结果
目标化合物对二斑叶螨卵的生物活性测试见表1。

表1 化合物I4a-I4e对二斑叶螨卵的卵孵化抑制率
目标化合物对二斑叶螨卵的室内生物测试实验表明,该系列目标化合物在质量浓度为100 mg/L时,对二斑叶螨卵的孵化抑制效果明显,其中目标化合物I4e抑制率为100%,当浓度降为100 mg/L时,目标化合物对二斑叶螨卵的孵化抑制率出现了不同程度的下降,但化合物I4a、I4b、I4e卵孵化抑制率均在60%以上,目标化合物IV1e在10 mg/L时的卵孵化抑制率为59%。构效分析结果为R基团芳环取代在邻对位活性高于在间位,如I4b>I4c。
2.2 合成条件影响
在合成中间体溴代丙酮酸乙酯时,使用不同的溴代试剂会造成不同的反应结果,在使用溴素为溴代试剂时,反应高效,在使用N-溴代丁二酰亚胺为溴代试剂时,反应进行不彻底,且在后处理中会产生大量的废水,因此合适的溴代试剂为溴素,另外在溴代时,使用二氯甲烷为溶剂进行溴代,反应速度快于以乙酸乙酯、二氯乙烷为溶剂,所以此反应的最佳条件为以二氯甲烷为溶剂,使用溴素进行溴代。
3 结论
以噁虫酮和噻虫嗪为先导化合物,从2,6-二氟苯甲酸出发,经氯代、胺解、硫代、缩合等反应,设计合成了一系列含1.2.4-噁二唑噻唑类化合物,并对所合成的化合物进行了1H NMR13C NMR ESI HRMS 的表征,并对所合成的化合物做了杀二斑叶螨卵的室内生物活性测试,结果表明,该系列化合物在质量浓度为500 mg/L时,对二斑叶螨卵的孵化抑制具有较好活性,其中化合物I4e抑制率为100%,在质量浓度为100 mg/L时,化合物I4a、I4b、I4e的卵孵化抑制率仍能达到60%以上,可作为新的先导化合物为后续研究提供参考,构效关系为R基团芳环取代在邻对位时活性高于在间位,并对中间体的合成工艺做了探索,溴代丙酮酸乙酯最佳的合成方法为以二氯甲烷为溶剂,使用溴素进行溴代。
符号说明
1.TLC,Thin Layer Chromatography,薄层色谱。
2.m.p.,melting point,熔点,单位:℃。
3.1H NMR,Proton Nuclear Magnetic Resonance,核磁共振氢谱。
4.13C NMR,Carbon Nuclear Magnetic Resonance,核磁共振碳谱。
5.J,Coupling Constant,耦合常数,单位:Hz。
6.δ,Chemical Shift,化学位移。
7.HRMS,High Resolution Mass Spectrometry,高分辨率质谱。
