Hermansky-Pudlak综合征基因型和临床表型分析
2022-11-18杨尚英程婉玉张焱盛迅伦
杨尚英 程婉玉 张焱 盛迅伦,3
1宁夏回族自治区人民医院 宁夏眼科医院,银川 750001;2宁夏医科大学科技中心电镜室,银川 750001;3甘肃爱尔眼视光医院,兰州 730000
Hermansky-Pudlak综合征(Hermansky-Pudlak syndrome,HPS)(MIM 203300)是白化病综合征中的一种,呈常染色体隐性遗传,具有明显的遗传和临床表型异质性。HPS具有11种亚型,分别为HPS-1~11,其中HPS-1和HPS-3为常见亚型,HPS-5较少见[1-3]。HPS临床表现多样,以眼皮肤白化病(oculocutaneous albinism,OCA)、出血倾向和组织内蜡样脂质聚积三联征为主要特征,伴或不伴致命性并发症,如肺纤维化、肉芽肿性结肠炎、肾衰竭及心肌病等。这些特征是由于溶酶体相关细胞器的缺陷,如黑素细胞中的黑素小体和血小板中的血小板致密颗粒缺乏或大量减少所致。目前,人类基因突变数据库(Human Gene Mutation Database,HGMD)已收录二百余种HPS基因变异。不同HPS基因编码的蛋白属于不同的复合体,具有不同的作用机制;而影响同一细胞器生物合成复合体(biogenesis of lysosome-related organelles complex,BLOC)或衔接蛋白-3复合体(adaptor complex-3,AP-3)的不同亚型往往具有相似临床表型,且不同亚型严重程度和临床表型不一,这为临床医生的鉴别诊断带来极大挑战。本研究采用全外显子组测序技术对2个HPS家系的致病基因进行筛查,并结合既往报道对HPS相关眼病的基因型及临床表型特点进行分析,分析总结此类疾病基因型与表型的关系。
1 资料与方法
1.1 一般资料
采用家系调查研究方法,纳入2020年6月至2021年5月在宁夏回族自治区人民医院就诊的中国汉族和回族HPS家系各1个,收集先证者及其父母的临床资料。本研究遵循《赫尔辛基宣言》,研究方案经宁夏回族自治区人民医院伦理医学委员会批准(批文号:2016018),所有基因检测及诊断工作均取得患者及家属的同意并自愿签署知情同意书。
1.2 方法
1.2.1家系成员临床检查 完善先证者及其父母的相关临床检查,包括面部外观照片、最佳矫正视力(best corrected visual acuity,BCVA)、裂隙灯显微镜、色觉、彩色眼底照相(TRC-NW300,日本Topcon公司)、光相干断层扫描(optical coherence tomography,OCT)(HD-OCT4000,美国Carl Zeiss Meditec公司)、视网膜电图(electroretinogram,ERG)、视觉诱发电位(visual evoked potential,VEP)和荧光素眼底血管造影(fluorescein fundus angiography,FFA)检查,并进行实验室凝血功能及肺部CT检查。
1.2.2电子显微镜下观察先证者血小板致密颗粒 采用含有枸橼酸钠和葡萄糖溶液的采血管采集受试者外周静脉血4 ml;离心半径12 cm,1 500 r/min离心10 min,静置后得到富含血浆的血小板,包埋法获取血小板切片;采用透照法观察血小板和血小板致密颗粒形态变化。电子显微镜下观察发现血小板致密颗粒缺乏或大量减少是诊断HPS的金标准[4]。
1.2.3家系成员全外显子组测序 先证者及其父母同时进行全外显子组测序(Trio全外显子组测序模式)。采用Agilent SureSelect外显子捕获试剂盒进行全基因组外显子捕获,采用高通量测序仪(美国Illumina公司)进行测序。采用Illumina basecalling Software 1.7分析软件对原始测序数据进行处理,并与NCBI人类基因组DNA参照序列(NCBI build 37.1)进行比对,采用SOAP软件(http://soap.genomics.org.cn)分析得到单核苷酸变异相关信息,采用BWA软件(http://bio-bwa.sourceforge.net/)分析得到插入/缺失变异相关信息。根据测序深度、测得变异reads数与该位点总reads数的比例、是否在高GC区域、是否在重复序列区域、是否在poly区域等对测序数据进行质控;并明确表型基因是否共分离;对1KG、gnomAD等数据库频率进行筛选,利用超过20种生物信息学软件预测,从而判别变异所在基因与疾病的关系。经过逐步过滤,筛选出家系内所有患者共享的变异数,再过滤家系无患病亲属存在的变异,最后进行变异注释及危害预测,获得候选致病基因变异。对可疑致病变异进行Sanger验证及共分离分析,并对缺失变异补充PCR检测。
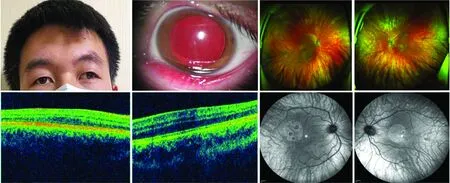
图1 家系1先证者(HPS-3型)临床资料 A:患者毛发、皮肤未见明显色素减退表现 B:左眼眼前节照相可见虹膜透光试验阳性 C、D:眼底照相可见双眼眼底色素缺失 E、F:双眼黄斑区OCT检查未见黄斑中心凹结构 G、H:荧光素眼底血管造影可见双眼弥漫性视网膜色素上皮层萎缩和脉络膜毛细血管萎缩,透见稀疏的脉络膜大血管 C、E、G示右眼,D、F、H示左眼Figure 1 Clinical data of proband with HPS-3 in family 1 A:No obvious depigmentation was observed in hair and skin B:Positive light transmission was found in the iris in anterior segment image of left eye C,D:Pigment deficiency was seen in both eyes E,F:Foveal structure was not found in both eyes in macular OCT images G,H:Diffuse retinal pigment epithelium atrophy,choriocapillary atrophy and visible sparse choroidal macrovascularity were detected in both eyes in fluorescein fundus angiography images C,E,G:right eye;D,F,H:left eye
1.2.4基因变异致病性分析 利用HGMD、dbSNP(https://www.ncbi.nlm.nih.gov/snp/)等数据库查询目标变异位点,在HGMD中查看是否为已报道的致病变异以及是否已被收录。如果是未报道过的新变异,依据2015年美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)发布的《序列变异解读标准和指南》对新发变异进行致病性评估。采用在线工具LRT(https://acronyms.thefreedictionary.com/Likelihood% 20ratio%20test)进行致病性预测。选用1000 Genome(http://browser.1000genomes.org/index.html)、EVS(http://evs.gs.washington.edu/EVS/)和ExAC(http://exac.broadinstitute.org/)数据库,查看变异在正常人群中的等位基因频率。
2 结果
2.1 2个HPS家系先证者临床表型
家系1中先证者父母近亲结婚。先证者,男,21岁,双眼外斜、视力差、畏光10余年,其面部皮肤、头发、眉毛、睫毛均无明显色素减退表现。双眼BCVA均为0.3(右眼:-1.50 DS/-2.00 DC×25°;左眼:-1.75 DS/-2.75 DC×155°),外斜-15°,水平眼球震颤,红绿色觉正常。双眼虹膜萎缩,透光试验阳性,余眼前节检查未见异常;双眼眼底呈橙色,色素缺失,可透见脉络膜大血管,OCT提示双眼黄斑中心凹发育不良(图1)。凝血功能检查仅凝血酶原时间轻微延长,为14.00 s。肺部CT检查未见明显异常。
家系2,先证者,男,4岁,其头发、眉毛呈棕黄色,全身皮肤颜色正常,双膝部可见少量淤青。双眼BCVA均为0.04(右眼:-4.00 DS/-5.00 DC×5°;左眼:-3.00 DS/4.00 DC×85°),色觉检查无法配合,双眼前节正常,眼底表现与家系1先证者相似(图2)。
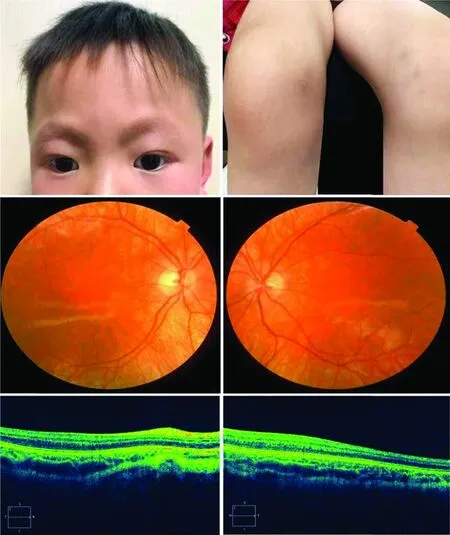
图2 家系2先证者(HPS-5型)临床资料 A:患者毛发、皮肤呈棕黄色 B:双膝可见少量淤青(箭头) C、D:眼底照相可见双眼眼底色素缺失 E、F:双眼黄斑OCT检查未见黄斑中心凹结构 C、E示右眼,D、F示左眼Figure 2 Clinical data of proband with HPS-5 in family 2 A:The hair and skin were brown yellow B:A small amount of bruises (arrow) were seen in both knees C,D:Pigment deficiency was seen in both eyes in fundus images E,F:Foveal structure was not found in both eyes in macular OCT images C,E:right eye;D,F:left eye
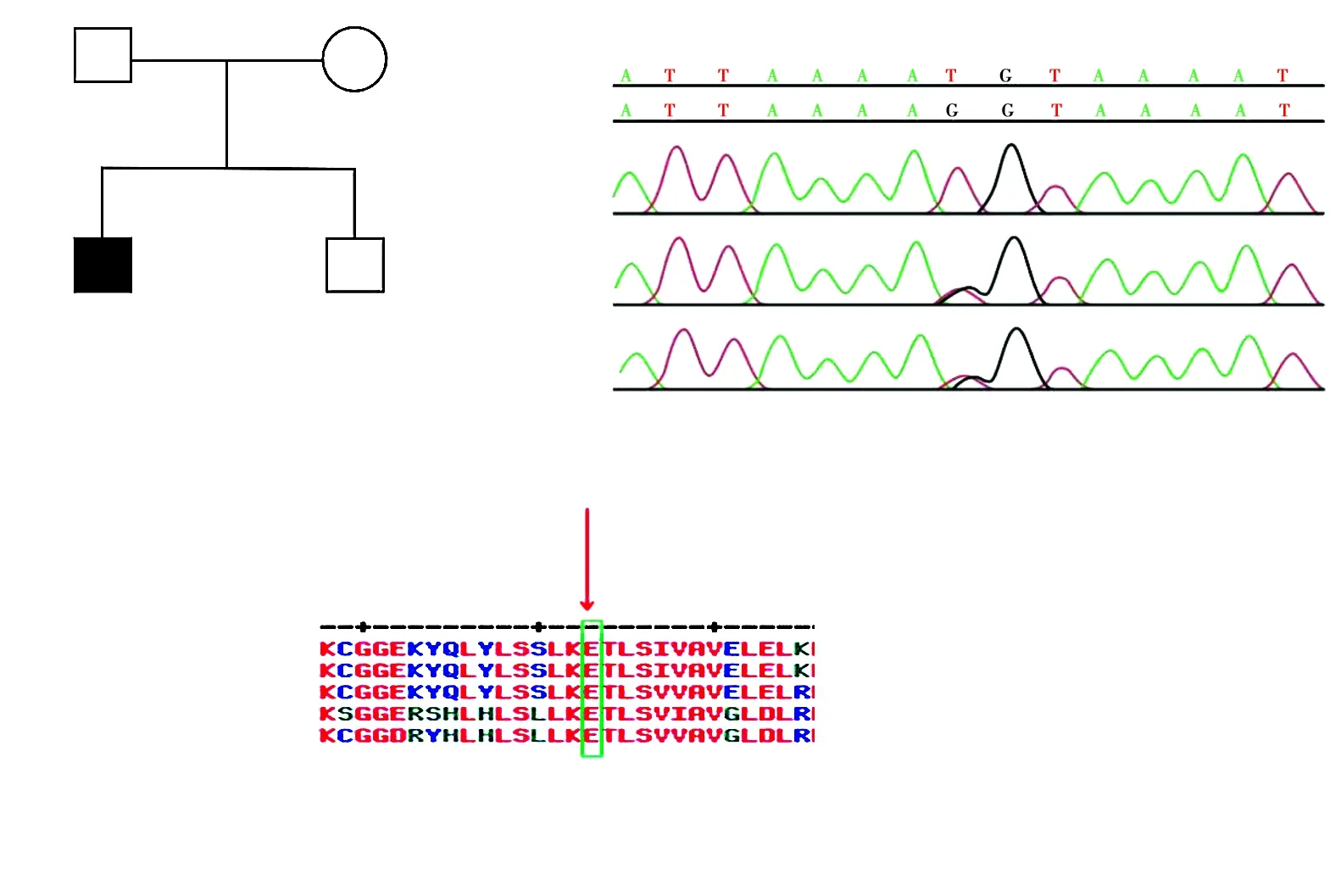
图4 家系1家系图及HPS3基因测序图 A:家系图 □:正常男性;○:正常女性;■:男性患者;:先证者 B:HPS3基因测序图 先证者携带c.2887G>T(p.E963X)纯合变异,父母各携带1个杂合变异 C:变异位点的氨基酸序列与多个物种的同源蛋白序列对比 963位氨基酸E属于高度保守序列Figure 4 Pedigree and sequence analysis of proband with HPS-3 in family 1 A:Pedigree of family 1 □:normal male;○:normal female;■:affected male;:proband B:Sequence chromatograms of HPS3 The proband Ⅱ-1 carried a homozygous variant of c.2887G>T(p.E963X),and the probnad's parents both carried a heterozygous variant C:The homology of amino acid sequences between human and other species The amino acid at position 963 (within box) was highly conserved among species
凝血功能检查仅凝血酶时间轻微延长,为19.90 s。肺部CT检查未见明显异常。
2.2 2个HPS家系先证者血小板致密颗粒形态学变化
电子显微镜下观察可见,家系1和家系2先证者均表现为血小板致密颗粒大量减少(图3)。
2.3 全外显子测序及生物信息学分析
家系1先证者HPS3基因第16外显子上检测到c.2887G>T(p.E963X)新纯合无义变异,表型正常的父母均携带c.2887G>T(p.E963X)杂合变异,提示基因型与临床表型共分离。该家系符合常染色体隐性遗传方式。结合患者临床表型,初步诊断为HPS-3型(图4)。家系2先证者HPS5基因上检测到复合杂合变异:c.2952-2A>C剪接变异和杂合缺失(缺失3 144 bp,位于chr11:18302108-18305251,exon22)。表型正常的父母分别携带1个杂合变异。Sanger测序验证剪接位点变异来源于父亲,缺失变异来源于母亲,符合常染色体隐性遗传方式,结合患者临床表型,初步诊断为HPS-5型(图5)。
2个家系确定的3种变异在HGMD均未收录,为新发变异。家系1先证者HPS3基因c.2887G>T变异导致多肽链提前终止合成,所产生的蛋白质大多失去活性或丧失正常功能(PVS1)。氨基酸保守性分析发现,HPS3基因翻译的氨基酸序列第963位谷氨酸高度保守(图4),改变后的氨基酸会导致蛋白结构功能变化而致病。变异与疾病在家系中共分离(PP1_Supporting)。变异在千人数据库和东亚人群数据库(ExAC_EAS)中未见报道(PM2_Supporting)。经过LRT和variant Taster软件预测,分别为有害变异和可能有害,具有致病性。根据ACMG指南,确定该变异为致病变异。
家系2先证者携带HPS5基因复合杂合变异c.2952-2A>C剪接变异和11号染色体杂合缺失(缺失3 144 bp,位于chr11:18302108-18305251,外显子22,PM2_Supporting)。c.2952-2A>C变异位于经典剪切区域,破坏了“GT-AG”剪切结构,推断会造成剪切异常,影响蛋白编码(PVS1)。2个变异在千人数据库和东亚人群数据库(ExAC_EAS)中均未见报道(PM2_Supporting)。变异与疾病在家系中共分离(PP1_Supporting)。根据ACMG指南,该变异判定为致病变异。
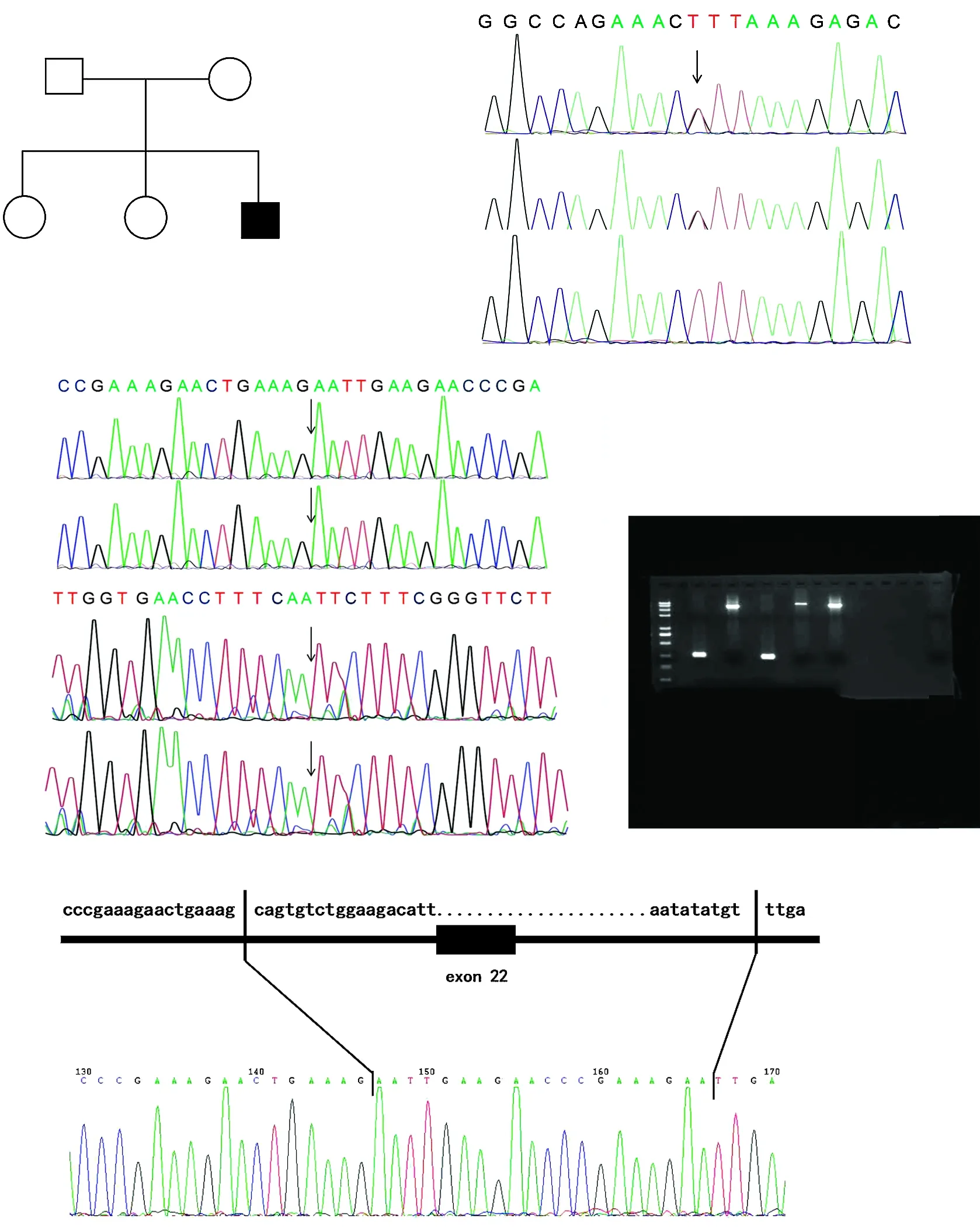
图5 家系2家系图及HPS5基因测序图 A:家系图 :正常男性;○:正常女性;■:男性患者;:先证者 B、C:HPS5基因测序图 先证者Ⅱ-3携带复合杂合变异,M1:c.2952-2A>C剪接变异和M2:缺失3 144 bp(包含外显子和内含子);先证者父亲Ⅰ-1携带M1杂合变异,先证者母亲Ⅰ-2携带M2杂合变异 D:PCR电泳图 短片段代表HPS5基因第22外显子的缺失,长片段代表野生型 E:断裂点图 断裂点染色体位置chr11:18302106和chr11:18305252,在缺失的146~166间插入了约20 bpFigure 5 Pedigree and sequence analysis of proband with HPS-5 in family 2 A:Pedigree of family 2 :normal male;○:normal female;■:affected male;:proband B,C:Sequence chromatograms of HPS5 The proband Ⅱ-3 carried a compound heterozygous variation,M1:c 2952-2A>C and M2:a 3 144-bp deletion (including exons and introns).The proband's father Ⅰ-1 carried the M1 heterozygous variation and the proband's mother Ⅰ-2 carried the M2 heterozygous variation D:PCR electropherogram The short segment represented the deletion of exon 22 in HPS5 gene,and the long segment represented the wild type E:Breakpoint pattern Breakpoints on chromosome positions chr11:18302106 and chr11:18305252,and about 20 bp was inserted between deleted 146 and 166
3 讨论
HPS3基因(NCBI RefSeq:NM_032383)定位于3q24,包含17个外显子,编码的HPS3蛋白包含1 004个氨基酸。HGMD中已报道39种HPS3基因变异,变异类型包括移码、重复、无义和错义变异[5]。HPS5基因(NCBI RefSeq:NM_181507)定位于11p15.1,包括23个外显子,编码含1 129个氨基酸的HPS5蛋白。HGMD中已报道37种HPS5基因变异,其中大部分是移码变异和错义变异[6-7]。HPS-5型发病相对少见,目前国内仅见2例报道,在HPS5基因上发现4种变异[7-8]。本研究在2个HPS家系中确定了3种新发变异。家系1为纯合无义变异(父母近亲结婚),家系2为剪接位点和杂合缺失的复合杂合变异。以上3个新发变异HGMD尚未收录,在正常人群数据库中的频率为0,软件预测结果表明为有害变异,ACMG遗传变异分类标准与指南评为致病变异。
HPS是一种罕见的异源染色体隐性遗传疾病,以溶酶体和溶酶体相关细胞器的异常为特征。HPS以眼皮白化病和出血倾向为特征,同时伴有全身各系统、器官的并发症。不同HPS亚型色素减退程度不同,如HPS-1型患者的毛发和皮肤颜色可随年龄增长而逐渐加深,HPS-3型患者的毛发和皮肤颜色可正常或表现为轻度色素减退。本研究中家系1先证者为HPS-3型,其头发、眉毛及皮肤颜色无明显色素减退表现。患者诉年幼时毛发呈棕黄色,随年龄增长头发颜色有所加深,而皮肤颜色未见明显改变。HPS-1型患者随年龄增长色素加深的情况是否也会出现于HPS-3型患者还有待进一步研究证实。Tsilou等[9]报道,HPS-3型患者相对HPS-1型而言,眼部临床表现较轻。本研究家系1先证者眼部及全身临床表现均较轻,既无严重的视力损伤,也无致命性的肺纤维化。HPS患者常伴有轻中度红绿色觉异常,但本研究中HPS-3型先证者色觉正常。Jardón等[1]报道64例HPS患者中,HPS-3型患者BCVA优于HPS-1型,外斜视在HPS-3型患者中更常见,而内斜视在HPS-1型患者中更常见。本研究家系1先证者表现符合外斜视。出血倾向是各型HPS患者的常见表型之一,多表现为皮下瘀血或皮肤易瘀青,严重出血可能发生于拔牙、手术或大的创伤后,女性患者可能伴月经量过多或产后出血,因此避免外伤及手术前做好相应止血措施,预防可能出现的大出血对HPS患者至关重要[10]。本研究2位先证者均具有出血倾向,这是由于血小板致密颗粒缺乏或大量减少导致血小板贮存池缺陷所致。血小板致密颗粒是一种溶酶体相关细胞器,富含H+、Ca2+、Zn2+等。血小板受到刺激活化后,血小板致密颗粒能够快速释放其内容物,如二磷酸腺苷酸等,使血小板聚集反应加强,还可引起血小板第二相聚集反应,从而促进凝血、血栓形成等多种生理学反应。近来,Yuan等[11]在研究中筛选到一种定位于血小板致密颗粒的锌离子转运蛋白TMEM163,小鼠缺失TMEM163会导致血小板致密颗粒内Zn2+的贮积和生物发生缺陷,并且TMEM163在BLOC-1、BLOC-2以及AP-3缺陷小鼠及HPS患者中均显著降低,表明这些复合体可能参与TMEM163运输到血小板致密颗粒的过程。不论是这些复合体,还是TMEM163本身的缺乏,都将导致血小板致密颗粒的发生缺陷,从而导致凝血功能障碍。这一发现揭示了血小板致密颗粒缺陷、血小板贮存池病和HPS中血小板致密颗粒缺乏的关键分子机制。本研究采用电子显微镜观察2个HPS家系先证者血小板,均发现血小板致密颗粒大量减少,获得了明确的确诊依据。此外,文献报道,HPS患者合并的肺纤维化显示出特发性肺纤维化的许多临床、放射学和组织学特征,且发病年龄轻,第1个发病高峰为20~25岁,高分辨率CT是诊断肺纤维化的金标准[2,12]。本研究中HPS-3型先证者未能行高分辨率CT检查,肺部CT检查提示双肺未见活动性病变,后续我们将继续对该患者进行随访,密切观察疾病的进展并预防并发症。不同HPS基因编码的蛋白属于不同的复合体,具有不同的作用机制;而影响同一BLOC或AP-3的不同亚型往往具有相似的临床表型。Di Pietro[13]等的研究也证实,HPS-3、HPS-5和HPS-6型的发病机制具有共同的生物学基础。例如,本研究中家系2先证者为HPS-5型,与HPS-3型具有较多相同的临床表型,如水平眼球震颤、外斜视、眼底色素缺失和黄斑中心凹发育不良,肺部CT均正常,血小板致密颗粒大量减少,凝血功能基本正常。同时也存在一些差异,如家系2先证者头发、眉毛颜色相对家系1患者有色素减退表现,而虹膜色素正常,视力损伤也较重。而不同HPS亚型患者之间的临床表型具有明显的异质性,如HPS-3、HPS-5型患者的主要特征是眼白化病(ocular albimism,OA)或OCA,同时伴有全身各系统的并发症,如出血倾向、肉芽肿性结肠炎等[14]。迄今为止,国内外尚无关于HPS-3和HPS-5型肺纤维化的报道。肺纤维化是HPS-1和HPS-4型患者常见的并发症,主要表现为肺实质及肺间质进行性不可逆性纤维化,最终可因呼吸衰竭而死亡。HPS-2型患者的常见并发症为中性粒细胞减少所致的反复感染,典型的临床表现是慢性粒细胞缺乏症。此外,属于不同复合体的各亚型并发症的严重程度亦有明显不同,如HPS-3、HPS-5和HPS-6型患者症状较轻,HPS-7、HPS-8和HPS-9患者症状较重,HPS-1和HPS-4患者症状严重程度中等[14]。
基因检测是确诊HPS的重要技术手段,利用基因诊断技术明确HPS亚型将有利于临床医师减少误诊的发生,积极干预患者潜在并发症。本研究中家系1先证者经临床检查及基因检测确诊为HPS-3型,在止血措施完备的情况下,顺利为家系1先证者进行了双眼斜视矫正手术;家系2先证者既往误诊为OCA,通过全外显子组测序,明确了家系2先证者为HPS-5型患者。
综上所述,本研究中家系1和家系3分别为HPS-3型和HPS-5型,两型之间存在一定的基因型和表型对应关系。全外显子组测序技术可以快速、准确地筛查HPS候选基因,明确临床亚型,有利于临床医师对HPS的早期诊断和对潜在并发症的精准干预。对于HPS患者而言,目前仍缺乏大样本量的系统研究,各亚型临床表型具有高度异质性,尚缺乏统一客观量化的评判标准。例如,眼底和毛发、皮肤色素缺乏程度的判断有一定主观性,且部分亚型患者的毛发颜色随年龄增长而发生变化;评估黄斑发育情况的OCT检查,部分患者年龄小无法配合或因缺乏固视能力无法采集;部分病例易误诊为OA或OCA等。因此,如何完善HPS临床表型的相关检查、制定客观量化的评判标准,如何通过基因检测手段提高诊断率是HSP研究的重要方向。建议对于OA和OCA患者常规行HPS基因筛查,评估出血风险及全身各器官、系统的情况,并尽可能行全外显子组测序,注意与HPS的鉴别诊断,以避免HPS的漏诊或误诊。
利益冲突所有作者均声明不存在利益冲突
作者贡献声明杨尚英:病例采集、文章撰写及修改;程婉玉:收集数据、分析数据;张焱:完成电子显微镜检查并提供影像资料;盛迅伦:负责研究设计、文章智力性内容的修改及定稿
志谢感谢北京同仁医院李杨教授、首都医科大学附属北京儿童医院袁业锋教授提供的电子显微镜实验方法和相关操作技巧
