基于外显子组测序的Axenfeld-Rieger综合征致病突变筛查
2022-11-18王琦刘鑫娜邵正波原慧萍
王琦 刘鑫娜 邵正波 原慧萍
哈尔滨医科大学附属第二医院眼科 哈尔滨医科大学心肌缺血教育部重点实验室 哈尔滨医科大学附属第二医院未来医学实验室,哈尔滨 150086
Axenfeld-Rieger综合征(Axenfeld-Rieger syndrome,ARS)是一组涉及虹膜和房角的罕见发育性遗传病,其在新生儿中的发病率为1/100 000~1/50 000[1],多为常染色体显性遗传,偶有散发病例报道,无明显种族和性别差异。眼部以角膜后胚胎环、虹膜前粘连和虹膜发育不良为特征,约50%的患者伴有青光眼。ARS患者也可伴有颌骨发育不良及牙齿、脐部、心脏发育异常等。但因发育异常涉及的部位和严重程度不同,患者临床表现差异很大,虹膜异常可以表现为轻度基质变薄、纹理异常、瞳孔移位,也可因基质缺损导致虹膜裂孔(即多瞳孔)和瞳孔变形,而眼部轻微异常且不伴有全身发育缺陷者极易被误诊或漏诊,因此明确的基因诊断不仅有助于指导优生优育、理解发病机制,也可为临床诊断提供参考,更有助于提示医生关注全身其他系统发育异常,早期干预,改善患者预后。编码转录因子的PITX2和FOXC1基因是目前已知的ARS致病基因,但由于ARS具有遗传异质性,仅有40%的患者可检出上述基因变异[1],因此针对已知致病基因进行Sanger测序的传统ARS致病突变筛查方法无法为所有患者提供遗传诊断。全外显子测序(whole exome sequencing,WES)可以快速、高效地检测基因组中对蛋白质结构有影响的外显子区变异,结合生物信息学手段能够迅速锁定罕见的可疑致病变异。本研究采用WES结合Sanger测序验证对ARS患者进行基因突变检测和突变致病性分析,旨在明确ARS诊断,为患者及其家系成员提供精准的临床诊疗和遗传咨询,为ARS的机制研究提供遗传学基础。
1 资料与方法
1.1 一般资料
采用家系调查研究方法,纳入2018年就诊于哈尔滨医科大学附属第二医院眼科的中国汉族眼前节发育异常一家系,详细询问患者病史及家族史。先证者就诊时10岁,该家系3代共15人,其中患者3例。本研究遵循《赫尔辛基宣言》,研究方案经哈尔滨医科大学附属第二医院医学伦理委员会批准(批文号:KY2019-231),参与本研究的家系成员或其监护人均对本研究目的知情并自愿签署知情同意书。
1.2 方法
1.2.1眼科及全身一般检查 参与研究的家系成员均接受专业眼科检查,采用标准对数远视力表检查最佳矫正视力;采用裂隙灯显微镜(SL-1E,日本拓普康公司)检查眼前节;采用非接触式眼压计(CT-80,日本拓普康公司)测量健康人眼压,采用回弹式眼压计(TA01i,芬兰iCare公司)测定患者眼压;采用裂隙灯图像系统(YZ5T,苏州六六视觉科技股份有限公司)行眼前节照相以记录眼前节及房角情况;采用儿童数字化广域成像系统(RetCam,美国科瑞公司)观察并记录患儿房角异常情况;采用数字眼底照相机(CR-2,日本佳能公司)行彩色眼底照相记录视网膜、脉络膜情况。同时进行全身一般检查,必要时进行专科检查。
1.2.2基因组DNA提取 采集配合基因分析的家系成员外周静脉血2~5 ml,置于EDTA抗凝管中于-80 ℃条件下保存备用。按照外周血基因组DNA提取试剂盒(DP304,北京天根生化科技有限公司)说明书提取基因组DNA。
1.2.3外显子组测序 使用Qubit测量先证者DNA样本浓度,琼脂糖凝胶电泳检查样本质量,合格后进行WES。使用Agilent SureSelect人全外显子V6试剂盒(美国Agilent公司)进行外显子捕获,文库构建完成后在NovaSeq 6000平台(美国Illumina公司)进行测序。
1.2.4原始数据处理及注释 过滤掉测序所得数据中带有接头和低质量的数据,得到的有效数据通过BWA软件比对到参考基因组(GRCh37/hg19),比对结果经SAMtools排序、Picard标记重复序列后,GATK软件检测单核苷酸多态性(single nucleotide polymorphism,SNP)和插入/缺失(insertion/deletion,InDel)变异,采用SnpEff软件对突变位点进行结构注释并过滤掉dbSNP中已经收录的等位基因频率>0.05的变异,结果文件经wANNOVAR工具[2]再次注释。

图1 ARS家系图 □:正常男性;○:正常女性;■:男性患者;●:女性患者;:先证者Figure 1 Pedigree of the ARS family □:normal male;○:normal female;■:affected male;●:affected female;:proband
1.2.5结合人群数据库和生物信息学评估筛选可疑致病突变 将注释后得到的SNP和InDel变异按如下步骤进行筛选:(1)保留剪接变异和外显子区变异,排除非编码变异和同义变异;(2)保留千人基因组计划(1000 Genomes Project,1000G)、Exome Aggregation Consortium(ExAC)、Genome Aggregation Database(gnomAD)基因组和外显子组测序数据库中全球或亚洲人等位基因频率<0.000 1或未收录的变异;(3)比对课题组内部与该家系无血缘关系者WES变异数据,排除2人以上同时具有的常见变异;(4)保留SIFT、PolyPhen-2、MutaionTaster、CADD(Phred>20)预测为有害的基因突变;(5)比对查找已报道ARS相关基因;(6)其他相关突变的排查,包括通过相互作用基因库检索工具(Search Tool Retrieval of Interacting Genes,STRING)数据库[3-4]对可疑突变基因进行蛋白相互作用网络分析,寻找PITX2、FOXC1相关蛋白;通过注释、可视化和集成发现数据库(Database for Annotation,Visualization and Integrated Discovery,DAVID)在线工具[5]对可疑基因进行基因本体(gene ontology,GO)分析,寻找与PITX2和FOXC1在相同生物过程(biological processes,BP)注释条目下的基因。
1.2.6Sanger测序共分离验证及致病性评估 针对可疑致病突变设计特异扩增引物,采用PCR试剂金牌Mix(TSE101,北京擎科生物科技有限公司)在S1000TMThermo Cycler PCR仪(美国Bio-Rad公司)上扩增目的片段后,Sanger测序验证排除假阳性,并在家系成员中验证该突变在正常家系成员和患者中出现基因型与临床表型共分离。引物合成和Sanger测序均由擎科生物哈尔滨分公司完成,测序结果用Chromas软件对基因序列进行对比分析。利用UGENE软件[2,6-7]对人、黑猩猩、猕猴、犬、牛、小鼠、大鼠、鸡、斑马鱼9种动物进行氨基酸的保守性分析。补充查找韩国基因组计划(Korean Genome Project,Korea1K)、Exome Variant Server(EVS)数据库,查看该突变在人群中的等位基因频率。参照美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)遗传变异分类标准与指南对候选罕见变异进行评估[8]。
1.2.7实时荧光定量PCR法检测健康人及受检者淋巴细胞PITX2 mRNA表达 按照超纯RNA提取试剂盒(CW0581S,北京康为世纪生物科技有限公司)说明书提取家系中3例患者和2名健康人以及1名与家系无关的健康人外周血淋巴细胞mRNA,采用逆转录试剂盒(Transcriptor First Strand cDNA Synthesis Kit,瑞士Roche公司)逆转录成cDNA,利用NCSYB GREEN qPCR Master Mix(NC-BM601,哈尔滨纳川生物技术有限公司)在C1000TMThermo Cycler荧光PCR仪(美国Bio-Rad公司)上进行实时荧光定量PCR检测。PITX2正向引物序列为5'-GACATGTACCCAGGCTA TTCC-3',反向引物序列为5'-GGGTTGACGTTCATAG AGTTGA-3'。以GAPDH作为内参,正向引物序列为5'-GTCTCCTCTGACTTCAACAGCG-3',反向引物序列为5'-ACCACCCTGTTGCTGTAGCCAA-3'。反应条件:95 ℃预变性15 min;95 ℃变性10 s,56 ℃退火及延伸30 s,39个循环。采用2-ΔΔCt法计算PITX2 mRNA相对表达量。
1.3 统计学方法
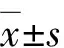
2 结果
2.1 家系成员临床特征
该家系中发病者在2代中连续出现,先证者双亲中有一方患病,符合已知ARS常染色体显性遗传特点,但不排除伴X染色体显性遗传可能(图1)。患者Ⅱ-3、Ⅲ-3和Ⅲ-4眼部表现为不同程度角膜混浊、角膜后胚胎环、虹膜发育不良、瞳孔移位、虹膜前粘连,伴双眼眼压升高(图2A~C)。其中先证者Ⅲ-3左眼角膜呈现灰白色混浊、虹膜隐约可见,激光扫描共焦显微镜检查发现左眼角膜上皮可见大泡、上皮下散在瘢痕结构、神经纤维缺失。3例患者均存在面中部扁平、牙齿缺损和尖牙、脐部皮肤突出(图2D~G)。Ⅰ-2、Ⅱ-4均未发现眼部及全身异常。
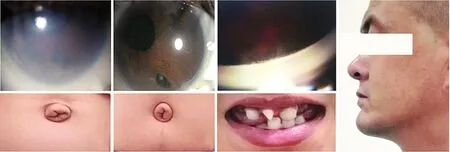
图2 Axenfeld-Rieger综合征患者临床特征 A、B:裂隙灯显微镜下可见患者Ⅲ-4双眼角膜混浊、后胚胎环(箭头)、瞳孔变形、移位、多瞳孔、虹膜发育不良(A:右眼;B:左眼) C:患者Ⅲ-4周边虹膜前粘连 D、E:患者Ⅲ-4和Ⅱ-3脐部皮肤突出 F:患者Ⅲ-3牙齿缺失、小牙 G:患者Ⅱ-3上颌骨发育不全伴面中部扁平Figure 2 Clinical features of patients with Axenfeld-Rieger syndrome A,B:Corneal opacification,posterior embryotoxon (black arrow),dyscoria,corectopia,polycoria and iris stroma hypoplasia in the right eye (A) and left eye (B) of patient Ⅲ-4 observed under a slit lamp microscope C:Peripheral anterior synechiae of patient Ⅲ-4 D,E:Protruding umbilicus of patients Ⅲ-4 (D) and Ⅱ-3 (E) F:Hypodontia and microdontia of patient Ⅲ-3 G:Maxillary hypoplasia with midface flattening of patient Ⅱ-3

图3 候选基因的生物信息学分析 A:已知致病基因FOXC1和候选基因的蛋白相互作用网络图(仅标注与其他蛋白有相互作用的候选基因蛋白名) B:与PITX2或FOXC1基因具有相同生物学过程的候选基因集合Figure 3 Bioinformatics analysis of candidate genes A:Protein-protein interaction network of FOXC1 and candidate genes (names of proteins diaplayed having interactions with others) B:Candidate gene set with the same biological processes of PITX2 and FOXC1
2.2 WES测序分析
先证者Ⅲ-3 WES目标区域覆盖度为97.74%,深度不低于30×的碱基比例为93.24%,各染色体平均覆盖深度均超过100×。排除测序结果中公共数据库和内部数据库非罕见变异,保留对蛋白质序列影响较大的突变121个。生物信息学软件预测发现,2个InDel变异、1个剪接突变、3个无义突变被MutationTaster评价为已知有害或可能有害,35个错义突变被CADD和SIFT评价为有害且MutationTaster和Polyphen-2至少1个软件评为有害,其中包括ARS致病基因PITX2的InDel变异。上述41个突变涉及的基因作为候选基因与已知ARS致病基因进行进一步生物信息学分析,STRING分析选取蛋白交互作用评分大于0.4(中等可信度)的基因,发现SIX2与PITX2相互作用(图3A),DAVID注释发现8个基因与PITX2或FOXC1具有相似功能,位于细胞迁移、DNA为模板的转录调控和起源于RNA聚合酶Ⅲ启动子的转录等GO-BP注释条目下(图3B)。
2.3 可疑致病突变验证及分析
上述候选基因变异中涉及ARS致病基因PITX2(NM_000325.6)杂合突变c.525delC,家系验证发现Ⅱ-3、Ⅲ-3、Ⅲ-4,即家系中临床诊断为ARS者均携带该突变,而表型正常的Ⅰ-2、Ⅱ-4不携带该变异,符合家系共分离(图4A)。3例患者外周血淋巴细胞中PITX2 mRNA相对表达量为0.672±0.063,明显低于健康对照的1.015±0.179,差异有统计学意义(t=8.847,P<0.001)(图4B)。dbSNP、1000G、gnomeAD、ExAC、Korea1K、EVS数据库中均未收录该变异,MutationTaster评为有害。预测蛋白质水平发生移码突变p.Asp175Glufs*(NP_000316.2),序列长度由324个氨基酸截短至206个,PITX2蛋白保守结构域之一OAR结构域缺失,受影响的氨基酸序列在9种动物中保守存在(图4C)。根据ACMG遗传变异分类标准和指南判定,该家系PITX2基因突变c.525delC/p.Asp175Glufs*满足1个PS证据、3个PM证据、2个PP证据,证实该突变为致病变异。

图4 PITX2基因突变Sanger测序结果及突变功能分析 A:患者及健康对照Sanger测序峰图 B:患者与健康对照PITX2 mRNA相对表达量比较 与健康对照比较,a P<0.001(独立样本t检验,n=3) C:PITX2基因p.Asp175Glufs*(c.525delC)突变与在蛋白质水平上发生移码突变及蛋白质截短、受突变影响的氨基酸序列在多种动物中保守存在Figure 4 Sanger sequencing result and functional analysis of the PITX2 mutation A:Sanger sequencing peak map of the patients and healthy family members B:Comparison of the relative expression of PITX2 mRNA between the patients and healthy controls Compared with healthy controls,a P<0.001 (Independent samples t test,n=3) C:The mutation p.Asp175Glufs*33 (c.525delC),subsequent PITX2 protein frameshift mutation and truncation,as well as the amino acid sequence affected were conservative
3 讨论
本研究首次在中国人中报道PITX2基因移码突变c.525delC(p.Asp175Glufs*),证实PITX2致病突变与ARS发病相关。在眼部,该基因突变除与ARS相关外,还与虹膜发育不良、虹膜房角发育异常综合征、Peters异常、角膜皮样环相关[9-11],PITX2致病基因突变也见于无眼部表现的牙齿发育异常者、先天性心脏病或房颤患者[12-15]。PITX2突变引起的表型多样性原因可能包括:(1)ARS患者也可出现牙齿和心脏异常,因此表型的不完全外显可能是导致临床差异的原因;(2)各患者的遗传背景有差异,如体内其他基因SNP的存在改变了患者对某些疾病表型的易感性,进而造成表型差异。在已报道的ARS家系中,患者眼部表型几乎全部外显,本研究中患者也均出现眼部异常及相似的牙齿和脐部发育缺陷,疾病累及部位无差异,仅严重程度表现不同。然而在个别ARS家系中,具有相同PITX2基因突变的患者疾病累及部位不尽相同[16],推测在同一家系中出现的表型异质性可能与患者间的遗传背景差异有关。因此,在ARS遗传学研究中,除关注疾病的致病基因外,获得ARS家系患者其他基因变异数据、分析家系的遗传背景差异,有望揭示ARS患者表型异质性原因及PITX2致发育异常机制。
目前进行大量基因序列平行测序的高通量测序技术主要为WES、全基因组测序和靶向基因测序(target gene sequencing,TGS),其中TGS和WES广泛用于单基因遗传病的变异筛查[17]。TGS也称panel测序,仅可检测目标基因片段变异,且panel的选择依赖于患者的临床诊断[18],而WES可以弥补这一点,适用于致病基因未全部明确的疾病。本研究利用WES技术除发现已知ARS致病基因PITX2变异外,也在其他基因中发现一些可能对蛋白质结构产生影响的罕见突变,涉及的基因与已知致病基因PITX2或FOXC1具有相似基因功能,其中SIX2基因编码蛋白与PITX2蛋白相互作用,但目前这些突变的致病性未知,需要进一步开展以家系为基础的遗传学分析和突变功能研究,以探讨这些候选基因与ARS发病和表型差异的相关性。
ARS与其他眼相关遗传病一样,除具有基因座异质性外,也具有等位基因异质性[17],ARS相关PITX2基因突变在形式上主要为截短突变(无义突变或移码突变)和重要功能结构域的错义突变,目前已报道的变异种类为100余种,其中错义突变和无义突变约45种,碱基插入或缺失约23种,约4种突变位于剪接区,但PITX2基因突变的形式和位置与患者表型严重程度尚未发现明显相关性,但与携带FOXC1基因突变的ARS患者表型相比,携带该基因突变的患者颅面部、牙齿及肚脐等全身发育异常更为常见[1]。本研究发现的c.525delC(p.Asp175Glufs*)移码突变改变了脊椎动物中保守存在的氨基酸序列,并导致PITX2蛋白末端及其内的OAR结构域缺失,而PITX2蛋白末端的OAR结构及其侧翼共39个氨基酸与PITX2蛋白的DNA结合和转录激活能力相关[19]。因此,c.525delC杂合突变可能作为功能获得性或功能缺失性基因突变改变PITX2蛋白功能,或因翻译形成的突变蛋白具有显性负性突变效应抑制野生型PITX2蛋白功能而致病,也可能通过移码突变介导的mRNA降解致PITX2基因单倍剂量不足引起ARS。针对患者体内PITX2基因mRNA表达水平检测发现,携带该变异的ARS患者PITX2 mRNA表达减少,且生物信息学预测该突变经转录翻译后蛋白质序列减少118个氨基酸(截短约36%),因此突变DNA链转录形成的mRNA被降解的可能性大,但最终能否翻译成功能蛋白及其对机体的影响仍需进一步研究。
综上所述,本研究利用WES技术结合Sanger测序在中国北方ARS一家系中发现了PITX2基因致病杂合突变c.525delC(p.Asp175Glufs*),该变异为首次在中国ARS家系中报道。本研究明确了该家系患者临床及遗传诊断,有助于提供符合疾病特征的诊疗方案,扩展了中国人PITX2基因突变谱,并强调了PITX2在ARS形成和发育相关遗传病发生中的作用。
利益冲突所有作者均声明不存在利益冲突
作者贡献声明王琦:设计试验、实施研究、采集数据、统计分析、论文撰写;刘鑫娜:实施研究、采集数据、统计分析、论文修改;邵正波:指导设计试验、论文修改;原慧萍:提供病例、指导设计试验、论文修改及定稿
