阿伐那非的合成工艺研究
2022-11-04梅玫曹棋程凤毛婕阳静丽胡华南
*梅玫 曹棋 程凤 毛婕 阳静丽 胡华南,2*
(1.九江市生态环境局 江西 332001 2.九江学院化学化工学院 江西 332005)
1.前言
阿伐那非(Avanafil,1)是由日本田边三菱制药株式会社授权美国Vivus公司开发,在2012年4月27日经美国FDA批准在美国上市,商品名为Stendra[1],主要用于治疗男性勃起功能障碍(ED)的药物。该药是一种口服速效的高选择性磷酸二酯酶-5(PDE-5)抑制剂[2-3]。与其他PDE-5相比,阿伐那非选择性更高,起效更快,副作用更少[4-5],预计将会有非常大的市场前景。
目前阿伐那非的合成路线如下:
路线一:苏贤斌等报道的阿伐那非的合成方法是一种新的固相合成方法,Merrifiled树脂与硫脲反应得到硫脲树脂,硫脲树脂依次经环合、氯代、取代、水解、缩合、氧化,最后与L-脯氨醇反应得到阿伐那非[6-7]。该路线Merrifiled树脂价格昂贵。
路线二:程晓峰等报道的阿伐那非的合成路线是以4-羟基-2-甲硫基嘧啶-5-羧酸乙酯为起始原料,依次经氯代、取代、水解、缩合、氧化,最后与L-脯氨醇反应得到阿伐那非[8-9]。该路线最后引入手性片段,也避免了手性中心消旋的风险,反应总收率为21.6%。
路线三:李志强等报道的阿伐那非合成路线是以甲基硫脲和乙氧亚甲基丙二酸二乙酯为起始原料经环合得到4-羟基-2-甲硫基嘧啶-5-羧酸乙酯,再依次经氯代、取代、氧化得到4-(3-氯-4-甲氧基苄胺基)-2-甲磺酰基嘧啶-5-羧酸乙酯,再L-脯氨醇发生亲核加成,水解后与2-氨甲基嘧啶盐酸盐缩合得到阿伐那非[10]。
课题组参考路线三[10-11],以4-氯-2-甲硫基嘧啶-5-羧酸乙酯(2)与3-氯-4-甲氧基苄胺(3)为起始原料,其发生亲核取代反应得到4-(3-氯-4-甲氧基苄胺基)-2-甲硫基嘧啶-5-羧酸乙酯(4),化合物(4)与间氯过氧苯甲酸(mCPBA)发生氧化反应得到4-(3-氯-4-甲氧基苄胺基)-2-甲亚磺酰基嘧啶-5-羧酸乙酯(5),化合物(5)与L-脯氨醇(6)发生亲核加成反应得到化合物(7),最后化合物(7)水解后与2-氨甲基嘧啶盐酸盐(9)发生缩合反应得到阿伐那非(1),合成路线见图1。
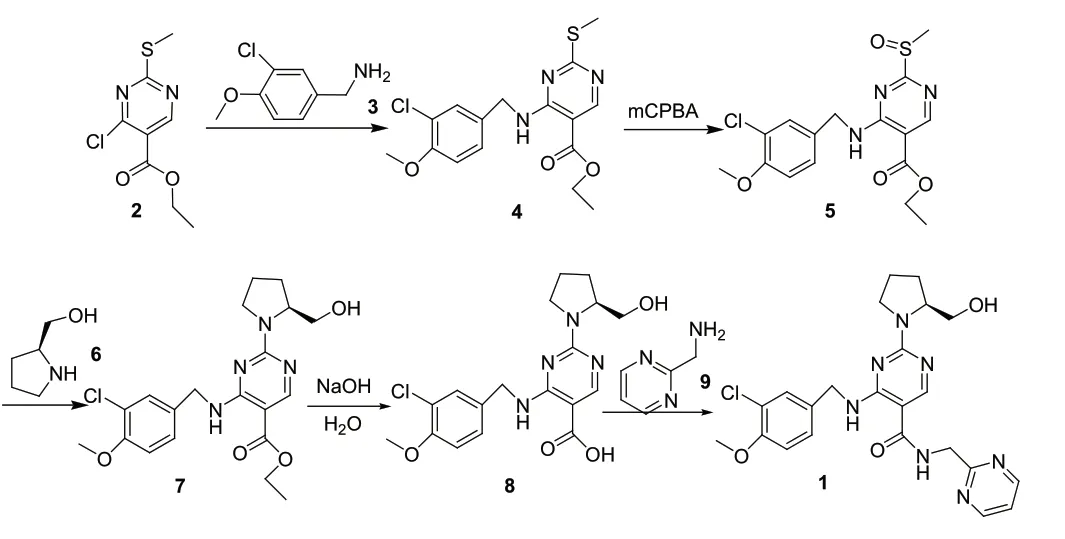
图1 阿伐那非(1)的合成路线
2.实验部分
(1)仪器和试剂
仪器:北京福凯科仪科技有限公司XT-4A型显微镜熔点测定仪(温度计未校正)、Bruker-300型核磁共振仪(四甲基硅烷作内标),Bruker Vector55型红外分析仪、Finnigan DECAX-30000 LCQ DecaXP plus电喷雾串联质谱仪、ShimadzuLC-10AT型高效液相色谱仪。
试剂:所用试剂均为CP或AR。
(2)分析方法
阿伐那非检测色谱条件:色谱柱:安捷伦ZORBAX-C18 250×4.6mm,5μm;柱温:30℃;进样量:10μL;样品浓度:1mg/mL;流速:1.0mL/min;检测波长:245nm;流动相A为0.1%三氟乙酸水溶液,B为乙腈,流动相比例:V(流动相A):V(乙腈)=40:60。
(3)实验步骤
①化合物(4)的合成
将20.0g(85mmol)4-氯-2-甲硫基嘧啶-5-羧酸乙酯(2)、70mL N,N-二甲基甲酰胺(DMF)置于反应瓶,冰水浴降温至0℃以下。将15.5g(90mmol)3-氯-4-甲氧基苄胺(3)、9.5g(94mmol)三乙胺及20mL DMF混合后滴加到反应液中,控制温度5℃以下。滴加完毕后,移去冰浴,升温至室温搅拌反应约1h。TLC确认反应结束后,将反应液倾入200mL 2%柠檬酸水溶液中,搅拌30min。用300mL乙酸乙酯(EA)分3次萃取水层至无产物,合并有机层。用200mL 10%柠檬酸水溶液洗涤2次,水洗,饱和食盐水洗涤,无水硫酸镁干燥,过滤,蒸干后溶于200mL乙酸乙酯中,加入100mL石油醚,0℃下搅拌析晶,过滤,真空干燥得28.0g淡黄色固体4,收率约94%,m.p.73~75℃;1H NMR(300MHz,CDCI3)δ:8.68(s,1H),8.60(brs,1H),7.37(d,J=2.0Hz,1H),7.21(dd,J1=8.4,J2=2.0Hz,1H),6.90(d,J=8.4Hz,1H),4.68(d,J=6.0Hz,2H),4.31(q,J=7.2Hz,2H),3.91(s,3H),2.51(s,3H),1.36(t,J=7.2Hz,3H);IR(KBr,v/cm-1):3321,2980,1680,1520,1256.ESI-MS(m/z):368[M+H]+。
②化合物(5)的合成
将13.8g(37.5mmol)化合物4和二氯甲烷(DCM)1500mL置于三口瓶,冰水浴降温至0℃,搅拌下,7.5g(43.5mmol)间氯过氧苯甲酸(mCPBA)分3批加入反应液中,控制温度0℃左右。加完升温至室温搅拌3h,TLC确认反应结束后,加入饱和碳酸氢钠溶液洗涤2次,水洗,饱和食盐水洗涤,无水硫酸镁干燥,过滤,旋蒸得到产物5,超过理论重量,m.p.66~68℃;1H NMR(300MHz,CDCI3)δ:8.65(s,1H),8.58(brs,1H),7.35(d,J=2.1Hz,1H),7.20(d,J=3.6Hz,1H),6.87(d,J=8.6Hz,1H),4.66(d,J=6.2Hz,2H),4.29(q,J=7.0Hz,2H),3.90(s,3H),2.52(s,3H),1.35(t,J=7.2Hz,3H);IR(KBr,v/cm-1):3325,2960,1682,1525,1250.ESI-MS(m/z):384[M+H]+。
③化合物(7)的合成
将14.5g(37.5mmol)化合物5置于三口瓶中,加入115mL的四氢呋喃(THF),常温搅拌,将4.2g(41.5mmol)L-脯氨醇(6)、4.6g(45mmol)三乙胺及30mL四氢呋喃混合均匀后,常温下滴加入反应液中,滴加完毕,常温搅拌反应,TLC确认反应结束后,蒸干溶剂。残余物溶于15mL乙酸乙酯(EA)中,饱和碳酸氢钠溶液洗涤1次,水洗1次,饱和食盐水洗涤,无水硫酸镁干燥,蒸干溶剂,加入75mL的无水乙醇升温全溶后,降温至室温析晶,降温-20℃析晶过夜,过滤,真空干燥得到类白色固体7约14.3g,以上两步反应总收率约91%,HPLC纯度为99%,m.p.86~88℃;1H NMR(300MHz,DMSO)δ:8.53(q,J=6.4Hz,1H),8.48(d,J=6.0Hz,1H),7.42(d,J=4.4Hz,1H),7.29(d,J=8.4Hz,lH),7.08(d,J=8.4Hz,lH),4.8l(m,1H),4.55(t,J=7.6Hz,2H),4.21(q,J=6.8Hz,2H),4.13~4.08(m,1H),3.82(d,J=1.6Hz,3H),3.63~3.58(m,1H),3.51~3.45(m,2H),3.35~3.31(m,1H),2.02~1.83(m,4H),l.26(t,J=7.2Hz,3H);IR(KBr,v/cm-1):3340,1674,1525,1257,804;ESI-MS(m/z):421[M+H]+。
④化合物(8)的合成
将12.6g(30mmol)化合物7置于三口瓶,加入100mL的二甲亚砜(DMSO),室温搅拌溶解,再加入12mL 10%的氢氧化钠水溶液,室温搅拌,TLC确认反应结束后,将反应液倾入250mL 10%的柠檬酸水溶液,降温至室温析晶,析出白色固体8,过滤,滤饼用水洗涤,真空干燥得到11.2g白色粉末状固体8。产率95.1%,m.p.202~204℃;1H NMR(300MHz,DMSO)δ:8.37(s,1H),7.35(d,J=8.2Hz,1H),7.24(s,1H),7.04(d,J=8.2Hz,1H),4.52(s,2H),4.06(s,1H),3.78(s,3H),3.57(d,-J=10.4Hz,1H),3.45(s,2H),3.32(t,J=9.0Hz,1H),1.91(s,2H),1.82(s,2H);ESI-MS(m/z):393[M+H]+。
⑤化合物(1)的合成
将9.8g(25mmol)化合物8置于反应瓶,加入100mL的N,N-二甲基甲酰胺(DMF),室温下搅拌溶解,然后再加入4.35g(30mmol)2-氨甲基嘧啶盐酸盐(9),依次加入5.8g(30mmol)1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(EDCI)、4.5g(30mmol)1-羟基苯并三唑(HOBt)、3.6g(36mmol)三乙胺(TEA),室温搅拌,TLC确认反应结束后,把反应液倾入150mL饱和碳酸氢钠溶液中,用150mL乙酸乙酯分3次萃取至水层无产物,合并乙酸乙酯层,有机层依次用50mL饱和食盐水、50mL饱和碳酸氢钠溶液洗涤,无水硫酸镁干燥,蒸干溶剂后得到的粗品溶于50mL甲醇中重结晶,过滤,真空干燥得到11.0g阿伐那非(1),白色固体。总收率91.5%,HPLC纯度为99.6%,m.p.150~152℃;1H NMR(300MHz,DMSO)δ:9.14(d,J=24.9Hz,1H),8.74(d,J=4.9Hz,2H),8.51(d,J=6.2Hz,1H),7.37(t,J=4.9Hz,2H),7.26(d,J=7.1Hz,1H),7.06(d,J=8.5Hz,1H),4.56(d,J=5.8Hz,2H),4.49(d,J=5.9Hz,2H),4.10(s,1H),3.80(s,3H),3.61(d,J=9.9Hz,2H),3.54-3.39(m,2H),2.12-1.66(m,4H);13C NMR(75MHz,DMSO)δ:167.4,160.8,159.8,157.3,156.7,156.4,153.3,133.3,129.3,127.7,120.5,119.7,112.7,98.2,61.8,58.8,56.0,47.3,44.8,42.1,27.6,22.6;IR(KBr,v/cm-1):3429,3048,2925,1606,1509,1435,1366,1308,1247,1177,1078,1029,906,835,786,626;ESI-MS(m/z):484[M+H]+。
3.结果与讨论
化合物(8)与2-氨甲基嘧啶盐酸盐(9),在1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(EDCI)、1-羟基苯并三唑(HOBt)和三乙胺(TEA)缩合剂催化下发生缩合反应,得到阿伐那非(1),重点考察了反应得物料比和缩合剂的用量对收率的影响。
(1)物料比对收率的影响
表1所示,当n(8):n(2-氨甲基嘧啶盐酸盐)=1:1.1时,产物的产率最高,再增加2-氨甲基嘧啶盐酸盐的量对收率没有明显影响。2-氨甲基嘧啶盐酸盐需要过量,可能在反应过程中少量氨基会被氧化,所以2-氨甲基嘧啶盐酸盐需要稍微过量。
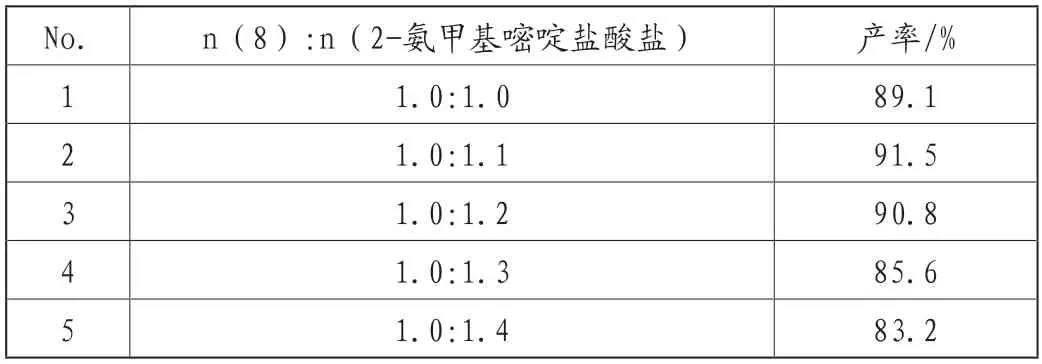
表1 物料比对阿伐那非(1)收率的影响
(2)缩合剂的用量对收率的影响
表2所示,缩合剂比例EDCI/HOBt/TEA=1:1:1.2时,当n(8):n(缩合剂)=1:1时,收率最高;随着n(8):n(缩合剂)从1:0增加到1:1.4,产物的产率先增加后减少,因为EDCI过多,产物会发生消旋,导致产率下降,同时EDCI和HOBt的用量必须相等,因为HOBt可以抑制消旋产物的产生。
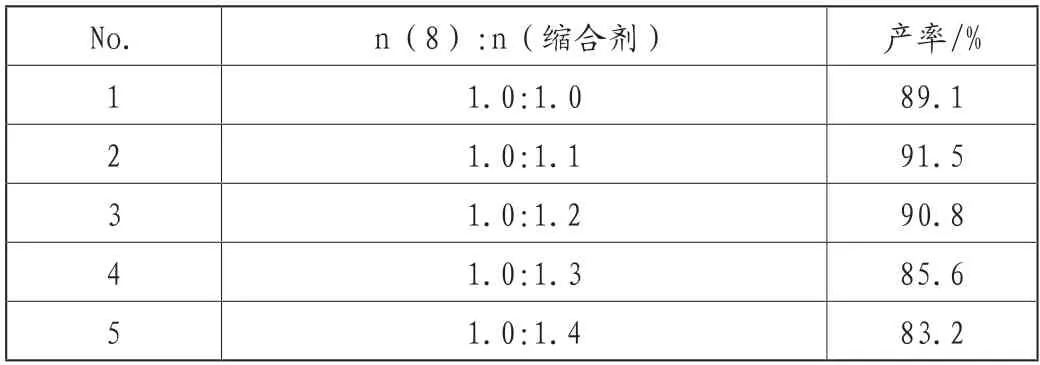
表2 缩合剂的用量对阿伐那非收率的影响
(3)谱图分析
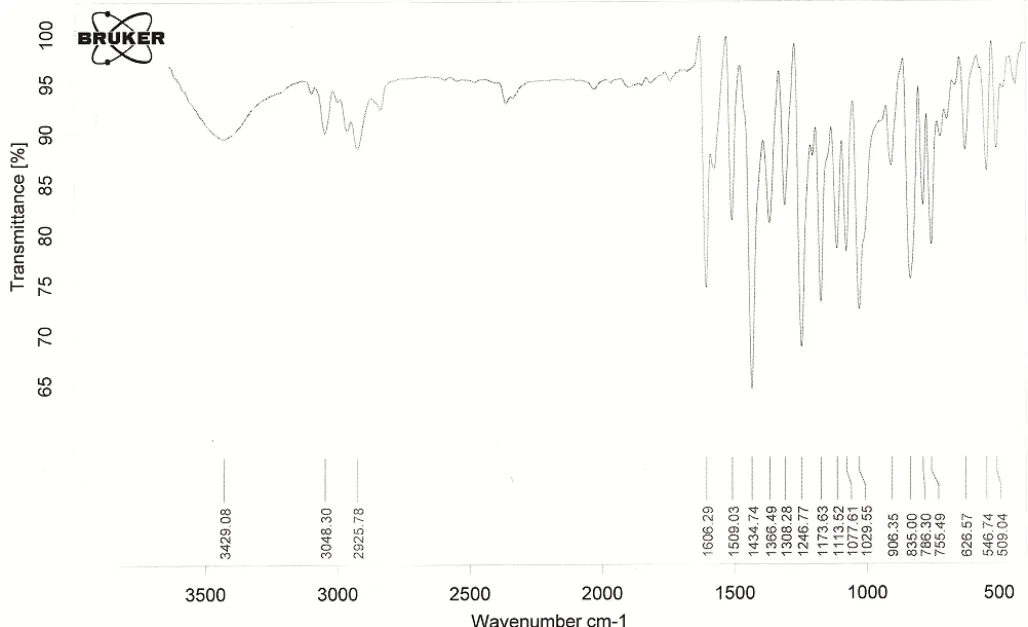
图2 阿伐那非(1)红外谱图
从IR的数据分析,在3429cm-1的吸收峰,对应为羟基的伸缩振动峰;在3048cm-1的吸收峰,对应为氨基的伸缩振动峰;在2925cm-1的吸收峰,对应为-CH2的对称伸缩振动峰;在1606cm-1的吸收峰,对应为C=O的伸缩振动峰。
4.结论
以4-氯-2-甲硫基嘧啶-5-羧酸乙酯(2)与3-氯-4-甲氧基苄胺(3)为起始原料,其发生亲核取代反应得到4-(3-氯-4-甲氧基苄胺基)-2-甲硫基嘧啶-5-羧酸乙酯(4),化合物(4)与间氯过氧苯甲酸(mCPBA)发生氧化反应得到4-(3-氯-4-甲氧基苄胺基)-2-甲亚磺酰基嘧啶-5-羧酸乙酯(5),化合物(5)与L-脯氨醇(6)发生亲核加成反应得到化合物(7),最后化合物(7)水解后与2-氨甲基嘧啶盐酸盐(9)发生缩合反应得到阿伐那非(1)。反应总收率达74.4%,产品经HPLC检测纯度达99.8%以上。
