改性纳米SiO2-环氧树脂浆料的制备及其性能研究
2022-09-15闫毅博王焕敏李雪飞牛利永李小红张治军
闫毅博, 王焕敏, 李雪飞, 牛利永,*, 李小红,, 张治军,*
(1. 河南大学 纳米材料工程研究中心,河南 开封 475004; 2. 河南河大纳米材料工程研究中心有限公司,河南 济源 459000)
近年来,随着电子信息产业的迅速崛起,电子产品持续朝向小型化、便携化、多功能化方向发展。在集成电路中,电子封装材料能有效分散有机基板和芯片间因热膨胀系数(Coefficient of Thermal Expansion, CTE)不匹配而产生在焊球表面的热应力,对芯片起到良好的支撑、保护及与外电路的连接作用,提高器件的使用可靠性[1]。灌封胶作为常见的电子封装材料,主要分为环氧树脂灌封胶、有机硅灌封胶以及聚氨酯灌封胶三类。由于芯片与基板间的间隙很小,要求灌封料的粘度极低,且具有良好的粘接性能和热膨胀稳定性[2]。环氧树脂灌封胶因其良好的耐高温性和绝缘性,以及对硬质材料优异的粘接力得到了广泛应用,但单纯的环氧树脂电子灌封胶存在渗透性弱、热膨胀系数较高等问题。目前主要解决方案是在环氧树脂灌封胶体系中引入低热膨胀系数的无机填料,例如硅微粉等,降低环氧树脂灌封胶热膨胀率,同时提高机械强度和介电性能[3]。
SiO2具有良好的耐热性、介电性能以及较低的热膨胀系数,其独特的球形结构使其在环氧塑封料中的添加量大幅提高,同时具有较小的应力集中[4],因而成为大规模集成电路基板和芯片封装主要填料。李禾、李朝阳、Guo等[5-7]均采用SiO2增韧改性环氧树脂;王建军等[8]以不规则熔融石英为原料,通过Ar-H2感应耦合等离子体技术制备球形硅微粉,经等离子体处理后可得到表面光滑、分散性好、球形度高的硅微粉,与原始粉末相比,球形硅微粉具有更高的填充量,当其填充量为75%(质量分数,下同)时,EMC的热膨胀系数显著下降。随着电子封装芯片尺寸的减小和输入/输出引脚密度的增加,为避免灌封胶在流动过程中微米级SiO2填料所带来的堵塞现象,对电子封装材料的填料提出了纳米化的要求[9-11]。但由于纳米SiO2具有较大的比表面积和过高的表面自由能,加入环氧树脂中后,极易出现团聚[12]、沉降、相分离等现象,以至于环氧塑封料(EMC)中纳米SiO2颗粒的特性难以得到有效的发挥。
基于上述问题,较为有效的解决方案是在有机相和无机相之间构筑良好的相容性界面[13-14],以提高SiO2在环氧树脂中的分散性[15],提升EMC的使用效果。
为了使填料在聚合物基体中稳定分散,科研工作者常采用化学改性对填料进行表面处理,化学改性方法又可以进一步分为硅烷偶联剂改性、醇酯改性及聚合物接枝改性,硅烷偶联剂改性因其原料成本较低,改性方法简单备受关注。Li等[16]利用不同官能团的有机硅烷偶联剂对SiO2纳米颗粒进行原位改性,研究了SiO2纳米填料的微观表面状态与复合材料的流变学、CTE和粘接性能之间的构效关系。但复合材料大多使用改性后的填料粉体与聚合物基体直接共混的方法制备而成[17-19]。采用该方法在制备复合材料的过程中,填料湿凝胶(表面未改性或者改性)的脱水干燥、煅烧过程不可避免会引起粉体的二次团聚[20-21],而硬团聚一旦形成就难以消除,导致粉体颗粒在聚合物基体中的分散变得尤为困难。因此本文探索在溶液的状态下,对表面改性后的SiO2进行共沸蒸馏,以最大限度去除SiO2表面羟基;同时,引入特征官能团,提高SiO2在环氧树脂中的分散性;溶剂状态下实现树脂与SiO2的有效复合,增加纳米SiO2在EMC中的填充量;充分发挥纳米微粒的特性,以制备在较高SiO2填充量下仍具有相对较低粘度以及良好的热膨胀稳定性的树脂浆料。
1 实验部分
1.1 仪器和试剂
JEM2100 PLUS透射电子显微镜(日本电子株式会社),ZS90激光粒度分布仪(英国MALVERN仪器有限公司),ZYMC-200(V)非介入式材料均质机(深圳中毅科技有限公司),Discovery HR-2旋转流变仪(美国TA仪器有限公司),Geminisem场发射扫描电子显微镜(德国卡尔蔡司公司),Q800动态热机械分析仪(美国TA仪器有限公司),Model 402 F1热机械分析仪(德国NETZSCH公司),TG 209 F3热重分析仪(德国NETZSCH公司),卤素水分测定仪MB35(美国OHAUS)。
无水乙醇、正硅酸四乙酯(TEOS)、氨水、3-缩水甘油醚氧丙基三甲氧基硅烷、正丁醇、丁酮等均为分析纯;环氧树脂-828(EP-828,济宁华凯树脂有限公司),甲基六氢苯酐(波林化工有限公司),1-氰乙基-2-乙基-4-甲基咪唑(四国化工有限公司),液体石蜡(西陇化工股份有限公司)。
1.2 样品制备
(1) 环氧基团修饰的纳米SiO2的合成(E-SiO2)
取适量的无水乙醇和氨水加入250 mL三口瓶中,待水浴锅温度升至55oC时,在机械搅拌350 rpm下,以0.5 mL/min的速率缓慢加入相应量的TEOS和无水乙醇的混合液,加料结束后反应1.5 h,将适量的3-缩水甘油醚氧丙基三甲氧基硅烷和等量的乙醇混合液逐滴加入三口瓶中,分别在60oC下反应1 h,80oC下反应2 h,然后加入一定量的正丁醇,在-0.07 MPa下进行溶剂置换,置换后的溶液在高温高压下进一步反应2 h,记为溶液A(溶液A中SiO2含量通过卤素水分测定仪进行测试)。
(2) 树脂浆料的制备
将适量的EP-828加入溶液A (加入EP-828的量以溶液A中SiO2含量计,SiO2在EP-828中的质量分数分别为10%、20%、30%、40%),搅拌30 min,直至EP-828完全分散,搅拌回流30 min,升温至100oC,在-0.07 MPa下减压蒸馏至溶剂几乎完全蒸出,将SiO2/环氧树脂浆料倒入表面皿内,在130oC的烘箱内干燥4 h,以去除未蒸干的正丁醇溶剂,最终得到具有不同填充量的SiO2-EP浆料。
(3) 复合材料的制备
称取相应计量比的固化剂甲基六氢苯酐和固化促进剂1-氰乙基-2-乙基-4-甲基咪唑于树脂浆料中(环氧树脂∶固化剂∶固化促进剂=1∶1∶0.01),在均质机内进行真空脱泡混料处理。将最终所制得的无气泡均匀混合物缓慢注入表面涂有液体石蜡的聚四氟乙烯模具中,在老化箱内150oC固化2 h,180oC固化2 h,冷却,得到固化后的复合材料。
2 结果与讨论
1.1 E-SiO2的表征
纳米SiO2的表面改性可以削弱表面极性基团间的相互作用,减轻团聚程度,提高SiO2的分散性。但即使是改性后的SiO2在脱水干燥情况下,也不可避免会发生二次团聚。图1为SiO2及E-SiO2分别在溶液和粉体状态下的SEM及粒径分布图。对比a1和b1,改性SiO2的分散性明显提高。经过干燥处理的SiO2发生了一定程度的团聚,平均粒径变大(c1和c2),这样不利于SiO2与树脂浆料的均匀混合。液相原位改性后的E-SiO2平均粒径约47 nm,较未改性的SiO2粒径(~50 nm)略有下降(a2、b2),表明环氧基团的引入可以限制SiO2颗粒的生长。通过在正丁醇溶剂中的高温高压处理,SiO2表面的羟基含量明显降低,由4.66×1020降至2.40×1020,这对进一步削弱SiO2表面极性,提高与树脂的相容性起到促进作用。考虑到E-SiO2干燥后会发生团聚,不利于与树脂浆料的共混,因此本文着重探索E-SiO2在溶液状态下制备树脂浆料并对其综合性能进行系统考察。
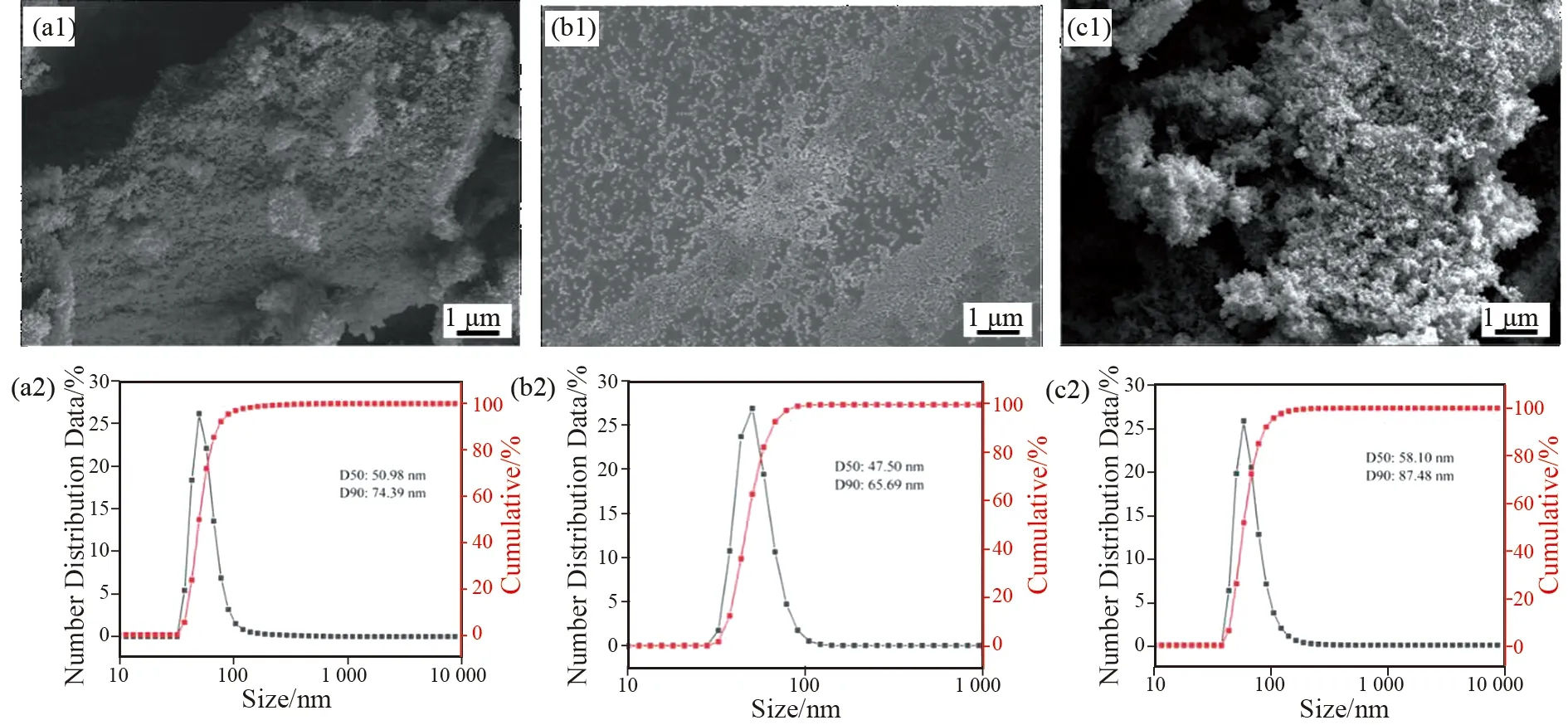
图1 不同状态下SiO2及E-SiO2的形貌及粒径表征(a)溶液状态下SiO2;(b)溶液状态下E-SiO2;(c)干燥状态下 E-SiO2
1.2 流变性能
粘度是EMC复合材料使用性能的关键指标。图2(a)和(b)分别为SiO2及E-SiO2在不同填充量下树脂浆料粘度随剪切速率的变化曲线图。由图2(a)中可知,纯EP在0.1~100 s-1的剪切速率下表现出牛顿流体的特性,粘度不随剪切速率变化。加入SiO2后,其树脂浆料的粘度随剪切速率的增加而下降,在SiO2填充量为30%,剪切速率在0.1~1 s-1之间,剪切减薄行为(剪切变稀)较为明显。主要原因是在外力的作用下,过量SiO2颗粒的加入导致聚合物链段的缠结点被打开,剪切速率越高,缠结点被打开的越多,原有的分子链构象发生变化,分子链沿流动方向,导致材料的粘度急剧下降[22-23]。

图2 不同填料填充量下树脂浆料粘度随剪切速率的变化曲线图(a) SiO2;(b) E-SiO2
对于E-SiO2树脂浆料,未施加剪切力时,体系中E-SiO2颗粒之间空隙较小,EP只能勉强填充其中的空隙,在较低的剪切力作用下,EP在其中起到了流体的作用,可作为E-SiO2颗粒之间的润滑剂,因此较同比例添加量的未改性SiO2初始粘度更低。当填料填充量为40%时,树脂浆料呈现出与其它样品完全不同的变化趋势:剪切速率为2~10 s-1时,粘度剧增,表现出剪切增稠效应,这是由于在剪切作用下,聚合物分子链间出现缠结行为,聚合物分子链与E-SiO2颗粒之间产生包覆和缠绕行为,均导致浆料粘度剧增;而当剪切速率增加至10 s-1以上时,较强的剪切力将聚合物分子链解缠结,导致浆料粘度急剧下降。
图3为改性前后SiO2在不同填料含量下树脂浆料的初始粘度对比图,相关的初始粘度值及降粘率列于表1。从表1可以看出,SiO2的填充量为10%,其树脂浆料初始粘度为92.7 Pa·s,E-SiO2树脂浆料初始粘度为90.0 Pa·s,降粘率仅为2.9%。结果表明:少量E-SiO2的加入降粘效果并不明显。当填料填充量逐渐增加,树脂浆料的初始粘度逐渐增加,这是由纳米SiO2较大的比表面积所导致的,SiO2在填充量达到40%时,干燥后的浆料已成块状,因此后续对复合材料断面、力学性能、动态热机械性能、热稳定性及热膨胀行为等的测试中,均未考察此样品。当填料的填充量为30%时,SiO2-EP浆料粘度为1 458.0 Pa·s,是纯EP粘度(10.5 Pa·s)的145倍,但E-SiO2树脂浆料粘度仅为371.1 Pa·s,与相同填料含量下SiO2的树脂浆料相比,降粘率达到74.5%。树脂浆料粘度下降主要是由环氧基团的引入降低了纳米SiO2的表面自由能,减弱了颗粒间的团聚所致。

图3 不同填料填充量下树脂浆料初始粘度对比图

表1 不同填料填充量下树脂浆料初始粘度及降粘率对比表
1.3 分散性
SiO2填料的分散性直接影响复合材料的综合性能。丁酮作为SiO2-EP浆料制备过程中常用溶剂之一,首先对SiO2-EP浆料在丁酮溶剂中的分散性进行了考察。图4为不同填料填充量的树脂浆料在丁酮溶液中的TEM图。SiO2-EP在丁酮分散液中的分散性较差,随着SiO2填充量的增加,聚集程度依次增加;但对于E-SiO2,随着填充量的增加,环氧树脂与E-SiO2间的界面相逐渐变的模糊,颗粒间保持良好的分散状态,这是由于环氧基团的引入降低了SiO2颗粒的表面自由能,使得粒子之间团聚倾向减弱所导致的。图5(a)为不同填料填充量的树脂浆料在丁酮溶液中的分散性光学照片,图5(b)为丁酮分散液的透光率随填料添加量的变化曲线图。从图5(b)可以看出,随着填料含量的增加,分散液的透光率逐渐下降;E-SiO2-EP浆料透光率均高于SiO2-EP浆料。当SiO2填充量为30%时,SiO2-EP浆料的纳米分散液透光率仅为3.1%,而E-SiO2树脂浆料的丁酮分散液透光率仍可以保持在50%以上,表明E-SiO2在环氧树脂中具有良好的分散性。此外,E-SiO2经过共沸蒸馏处理,减弱了硬团聚产生的可能,也有助于提高SiO2在环氧树脂中的分散性。因此E-SiO2的树脂浆料具有良好的透光率。

图4 不同填料填充量下树脂浆料丁酮分散液的TEM图(上:SiO2;下:E-SiO2)(a)10%;(b) 20%;(c) 30%;(d) 40%
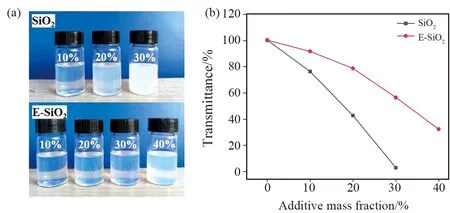
图5 不同填料填充量下树脂浆料的(a)分散性图片;(b)透光率
图6为日本竞品与本文所制备40%填料含量下的E-SiO2树脂浆料分散性的对比图。由图可知,二者在丁酮溶液中均具有较好的分散性,竞品SiO2的D50约为55 nm,本文产品的D50约为58 nm,二者均具有较窄的粒径分布。对比其透射图可知,本文所合成的产品粒径更为均一,球形度更好。二者的分散性对比结果表明本文所制备的产品与竞品相当。

图6 竞品与本文产品的(上:竞品;下:本文产品)(a)树脂浆料图片;(b)分散性图片;(c)激光粒径分布图;(d/e)TEM图
1.4 断面形貌
将制备的氧化硅/环氧树脂浆料固化,采用扫描电镜对其断面形貌进行考察,进一步评估填料在树脂体系中的分散性。图7为不同填料填充量下树脂浆料固化后的断面形貌图。由图可知,SiO2的填充量为10%时,并未出现明显的团聚现象,SiO2在环氧树脂中的分散性较好;当填充量逐渐增加,纳米微粒在溶液状态下极易发生硬团聚,在图中表现为明显的聚集体,分散性减弱。E-SiO2在环氧树脂中的分散性明显提高,这主要是由功能性环氧基团的引入降低了SiO2的表面自由能,减弱了颗粒间的相互吸引所致。

图7 不同填料填充量下树脂浆料的断面形貌图(上:SiO2;下:E-SiO2)(a)10%;(b) 20%;(c) 30%;(d) 40%
1.5 储能模量
不同填料添加量下固化后材料的储能模量(E’)随温度的变化曲线如图8所示。由图8(a)可以看出,在玻璃态区域,纯EP的E’为2 315 MPa;当加入10%的SiO2时,固化后材料的E’下降至1 715 MPa;原因是SiO2的引入改变了EP基体大分子间的相互作用,大量的硅羟基的引入影响了环氧树脂的固化过程,导致E’下降。而随着SiO2含量进一步增加,材料的E’反而提高,这是因为纳米SiO2粒子与环氧树脂间产生较强的相互作用,并起到物理交联的作用,使得聚合物链段运动能力减弱,有机-无机相间较强的物理作用足以抵消固化过程的影响[24]。对于E-SiO2,其与树脂间的相互作用由较弱的范德华力转变为较强的共价键相互作用,因此复合材料的E’均高于纯EP。当E-SiO2的添加量达到30%时,E’达到最大,为3 144 MPa;E-SiO2的含量继续增加,E’变化不大。改性与未改性的材料相比,显示出更高的E’,这表明界面相的性质对复合材料的动态热机械性能影响较为明显。
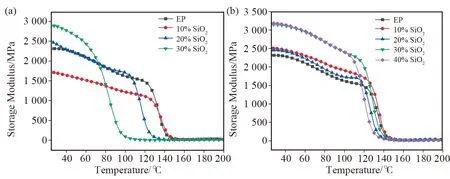
图8 不同填料填充量下树脂浆料的储能模量(a) SiO2;(b) E-SiO2
1.6 玻璃化转变温度
损耗因子为材料损耗模量与储能模量的比值,其随温度变化曲线的峰值所对应的通常用来表征玻璃化转变温度Tg。Tg决定了材料的工程应用条件。图9(a)和(b)分别为不同填料填充量下固化后材料的损耗因子随温度的变化曲线图,图9(c)为材料的玻璃化转变温度随填充量的变化曲线。由图可知,随着SiO2在环氧树脂中填充量的增加,Tg呈现出不同的变化规律。对于SiO2,Tg在10%的填充量时最高,相比纯EP,提高了约4 ℃;但随着SiO2填充量的继续增加,Tg急剧下降,远远低于纯EP的玻璃化转变温度,这是由于SiO2添加量较低时,环氧链段的运动受到阻碍,表现为Tg升高;当SiO2过量时,填料具有过大的比表面积和较强的吸附作用,使得EP大分子链运动进一步受限,并阻碍其在固化过程中的交联,因此固化后的EP大分子链自由体积增加,Tg下降[13]。对于E-SiO2,不同添加量下的材料玻璃化转变温度均低于纯EP。该现象已有相关报道[25-27],功能化纳米SiO2的塑化效应可以解释这一现象,塑化效应可能来自于功能化纳米SiO2表面的吸附水以及复合材料中残留的有机小分子[28]或纳米SiO2与树脂间的小间隙。当E-SiO2的添加量从10%增加至40%时,Tg始终维持在130 ℃以上,这主要是由于:1)均一分散的E-SiO2的加入对聚合物链段的运动起到限制作用;2)E-SiO2表面的环氧官能团可能参与了EP大分子链的固化过程,与大分子链形成了化学键合作用;二者均导致E-SiO2填充的复合材料具有较高的Tg。
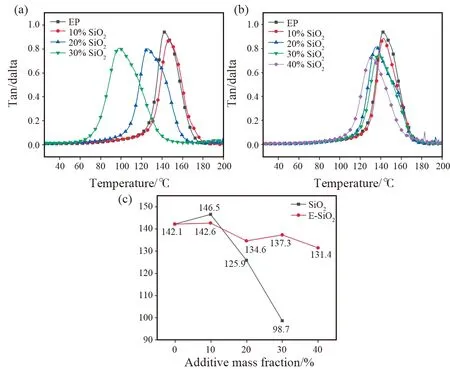
图9 不同填料填充量下树脂浆料的(a) SiO2损耗因子;(b) E-SiO2损耗因子;(c) Tg
1.7 热膨胀行为
决定EMC能否在电子封装领域使用的关键指标是热膨胀系数(CTE),较低的CTE有利于保持复合材料的尺寸稳定性。图10(a)和(b)为不同填料填充量下固化后的材料尺寸随温度的变化曲线图。由图可知,样品曲线斜率随温度的升高逐渐增加,但在100~150 ℃之间存在一个斜率变化较大的转折区域,通常认为该区域为复合材料的玻璃化转变区域,对复合材料在玻璃化转变前、后曲线的斜率进行计算,结果列于图10(c)和(d)。图10(c)为复合材料在玻璃化转变区域前(即玻璃态)的CTE值,图10(d)为玻璃化转变区域之后(即橡胶态)的CTE值,分别将其定义为CTE1和CTE2。如图所示,纯EP在玻璃态和橡胶态下的CTE值分别为65.2×10-6/K和188.3×10-6/K。当填料填充量增加至30%时,E-SiO2纳米复合材料的CTE1和CTE2分别为52.7×10-6/K和165.2×10-6/K,与纯EP相比,分别降低了19.1%和12.3%;E-SiO2含量继续增加,CTE1和CTE2均低于纯EP,这是由于E-SiO2参与树脂基体的交联,分子间作用增强,材料的热膨胀率降低, EMC的使用性能提高。

图10 (a) SiO2热膨胀行为;(b) E-SiO2热膨胀行为;(C) CTE1;(d) CTE2
3 结论
本文采用环氧硅烷偶联剂对二氧化硅进行液相原位改性;通过溶剂置换水热处理,降低表面羟基含量;溶剂状态下成功制备具有高SiO2填充量的环氧树脂浆料。环氧基团的引入降低了SiO2颗粒的表面自由能,减弱了颗粒间的相互作用力,E-SiO2在环氧树脂中分散性提高,树脂浆料的粘度降低,在丁酮分散液保持良好的透光率;E-SiO2通过化学键与环氧树脂结合,并参与树脂基体的交联,限制了聚合物链段的运动;当其添加量为30%时,复合材料的Tg较高,玻璃态和橡胶态下的热膨胀系数分别降低了19.1%和12.3%。该方法为在较高SiO2填充率下仍具有较低粘度的环氧塑封料的制备提供了有效途径。
