人源内皮抑素在大肠杆菌中的可溶性表达
2022-09-08张吉吴志勇朱玲玉辛瑜顾正华石贵阳张梁
张吉,吴志勇,朱玲玉,辛瑜,顾正华,石贵阳,张梁*
1(粮食发酵工艺与技术国家工程实验室(江南大学),江苏 无锡,214122)2(江南大学 生物工程学院,江苏 无锡,214122)
内皮抑素(endostatin,EDN)是一种抑制血管生成和肿瘤生长的内源性抑制剂,最初在小鼠内皮瘤细胞系的上清液中分离得到[1]。研究表明,EDN具有广泛的抗实体瘤活性的作用,具有抑制内皮细胞增殖、迁移、血管生成等功能,因此在血管增生性疾病的治疗中具有广阔的应用前景[2-4]。人源内皮抑素(human endostatin,hEDN)是人源胶原蛋白XVⅢ型C-端的184个氨基酸片段,分子质量约为22 kDa,其中酸性氨基酸16个,碱性氨基酸29个,半胱氨酸4个,疏水氨基酸占42%,等电点为9.3,与鼠源内皮抑素具有很高的同源性,两者约具有86%的相同氨基酸残基[5]。研究表明,hEDN 中的N末端的1,3,11三个组氨酸残基和76位的天门冬氨酸是4个Zn2+结合位点[6],hEDN与Zn2+结合对其抗血管生成活性具有很重要的作用[7]。
EDN作为一种功能性多肽,国内外对其进行了大量的研究。1999—2002年,由毕赤酵母表达的重组EDN在美国进行了Ⅰ期和Ⅱ期临床试验,其中Ⅱ期验证了其安全性,但是这两期研究的药效学结果不太理想。在I期临床试验中用EDN治疗皮肤伤口和肿瘤后无法检测到其血管的变化[8];并且在Ⅱ期临床试验中用EDN治疗肿瘤患者后发现并没有抑制肿瘤细胞的生长[9]。2001—2005年,国内对重组EDN进行了总共三期的临床试验,并通过在hEDN的N端增加氨基酸的方法来提高其可溶性和活性[10]。目前,EDN临床应用面临着溶解性低、稳定性差、价格昂贵、需要大剂量等问题[11]。因而,探索EDN的高效制备方法对拓展其在医药领域的应用具有重要意义。随着基因工程技术的发展,生物表达系统已被广泛应用于重组蛋白或多肽的表达和制备。目前,已有多种表达系统应用于EDN的表达,如哺乳动物细胞[12]、毕赤酵母[13]和大肠杆菌系统[14-15]。尽管哺乳动物细胞和酵母表达系统作为真核系统对真核细胞来源的蛋白或多肽的适用性更好,但成本较高且耗时长。大肠杆菌表达系统具有生长速度快、遗传背景好、表达水平高和价格低廉等优点,是生产重组蛋白较合适的系统之一[16]。乔路等[17]通过大肠杆菌(Escherichiacoli)实现了hEDN的原核表达,但是产物为包涵体。目前,虽然已经实现了hEDN在大肠杆菌中的表达,但是其产物多为蛋白质错误折叠而形成的不溶性包涵体,并且包涵体处理步骤复杂且复性成功率不高。因此,在大肠杆菌中探究hEDN的可溶性表达对实现其大量制备具有重要的意义。
融合标签在蛋白或多肽的表达过程中具有重要作用,如促进目的蛋白可溶表达,缓解异源蛋白毒副作用等。常用的融合标签包括谷胱甘肽巯基转移酶(glutathione S-transferase,GST)标签[18]、硫氧还蛋白A (thioredoxin A,Trx A)标签[19]、麦芽糖结合蛋白(maltose binding protein,MBP)标签[20]和小分子泛素样修饰蛋白(small ubiquitin-like modifier,SUMO)标签[21]等。值得注意的是,GST融合标签不仅是一种促溶标签,同时也是一种亲和蛋白纯化系统,通过在大肠杆菌中诱导表达GST融合蛋白,并通过将GST结合特性应用于谷胱甘肽来纯化融合蛋白[22]。研究表明,这些促溶标签能大大提高重组蛋白在大肠杆菌中的可溶性表达率[23]。因此,通过利用融合标签来促进异源蛋白的表达和生产是一种可行的策略。
本研究利用分子生物学技术分别构建了hedn单基因表达质粒、融合标签共表达质粒和融合表达质粒并转入到大肠杆菌中,摇瓶发酵并通过SDS-PAGE和串联飞行时间质谱(matrix-assisted laser desorption/ionization time of flight/ ionization time of flight,MALDI-TOF/TOF)鉴定hEDN的表达情况,进而探究hEDN的可溶性表达条件。进一步地,在优选的GST融合表达基础上,对摇瓶发酵的诱导温度、诱导时间、诱导剂浓度和添加诱导剂的时间等条件进行了优化,确定了hEDN融合蛋白的最佳发酵条件。
1 材料与方法
1.1 实验材料
1.1.1 菌株和质粒
本研究所用菌株和质粒见表1。

表1 本研究所用的菌株和质粒Table 1 Strains and plasmids used in this study
1.1.2 引物
本研究引物序列信息详见表2。

表2 本研究所用的引物Table 2 Primers used in this study
1.1.3 试剂与培养基
LB培养基(g/L):酵母粉5,胰蛋白胨10,NaCl 10,115 ℃灭菌20 min。
TB培养基(g/L):酵母粉24,胰蛋白胨12,甘油 5,KH2PO42.31,K2HPO412.54,115 ℃灭菌20 min。
主要试剂:限制性内切酶,美国Thermo公司;GoldBand 3-color Regular Range Protein Marker,上海Yeasen公司;T4DNA连接酶,大连Takara公司;重组PreScission Protease,上海源叶公司;2×TaqMaster Mix、2×PfuMaster Mix,杭州宝赛生物科技有限公司;质粒DNA提取、片段纯化、凝胶回收试剂盒,美国Axygen公司;PrePack GSH Purose 4 Fast Flow预装柱,江苏千纯公司;其他试剂均为国产分析纯。
1.1.4 仪器与设备
S100D型PCR仪、Chemi Doc凝胶成像仪,美国Bio-Rad公司;DYY-6C核酸电泳仪,北京六一仪器厂;Pico17高速离心机,美国Thermo Fisher Scientific公司;HYL-C型组合式摇床,太仓市实验设备厂;Sonic VCX-750型超声波细胞破碎仪,南京新辰生物科技有限公司;SCG蛋白纯化系统,苏州赛谱仪器有限公司;MALDI-TOF/TOF串联飞行时间质谱仪,美国Waters公司。
1.2 实验方法
1.2.1 人源内皮抑素表达质粒的构建
1.2.1.1 单基因表达质粒的构建
UniProt (www.uniprot.org)中查询得到人源内皮抑素氨基酸序列(Identifier:P39060),基于大肠杆菌密码子偏好性对其进行密码子优化,交由上海生工生物工程有限公司合成基因并将人源内皮抑素基因humanendostatin(hedn)序列克隆在pUC57质粒上,记为pUC57-hedn。以质粒pUC57-hedn为模板,利用PCR扩增片段,通过酶切、连接、转化、菌落PCR、测序等分子生物学手段,最终得到E.coliBL21(DE3)/ pET28a-hedn重组菌株。
1.2.1.2 融合标签共表达表达质粒的构建
构建方法同1.2.1.1,最终得到E.coliBL21(DE3)/pTIG-hedn重组菌株、E.coliBL21(DE3)/pETDuet-1-TrxA-hedn重组菌株、E.coliBL21(DE3)/pETDuet-1-hedn-TrxA重组菌株、E.coliBL21(DE3)/pETDuet-1-mbp-hedn重组菌株、E.coliBL21(DE3)/pETDuet-1-hedn-mbp重组菌株。
1.2.1.3 融合表达质粒的构建
构建方法同1.2.1.1,最终得到E.coliBL21(DE3)/ pET-32a-hedn重组菌株、E.coliBL21(DE3)/pGEX-6p-1-hedn重组菌株。
1.2.2 人源内皮抑素的诱导表达及质谱鉴定
将上述重组大肠杆菌分别划线于卡那霉素、氨苄青霉素抗性平板上37 ℃倒置培养12 h,挑取单菌落,接种于15 mL LB液体培养基中,37 ℃,200 r/min培养12 h,以1∶50体积比接种于50 mL LB培养基中,37 ℃,200 r/min培养至OD600=1.4~1.6时加入异丙基-β-D-硫代半乳糖苷(isopropyl-beta-D-thiogalactopyranoside,IPTG)至终浓度为0.2 mmol/L,在16 ℃,200 r/min条件下,诱导发酵24 h,12 000 r/min,5 min离心弃上清液收集菌体,用pH 8.0 PBS缓冲液(140 mmol/L NaCl,2.7 mmol/L KCl,10 mmol/L Na2HPO4,1.8 mmol/L KH2PO4)洗涤3次,将重悬后的菌体用超声破碎仪破碎,破碎条件:破1 s,停2 s,总时间24 min,4 ℃,12 000 r/min离心30 min,取上清液和沉淀,通过SDS-PAGE分析其表达情况。
将成功表达的SDS-PAGE凝胶条带切割后置于1.5 mL EP管中,然后对凝胶进行脱色、溶化、消化等预处理,利用MALDI-TOF/TOF分析。
1.2.3 人源内皮抑素诱导条件的优化
确定能成功表达hEDN的重组菌株后,使用TB培养基37 ℃,200 r/min条件摇瓶发酵24 h,每2 h取样,测OD600值,描绘该重组菌株的生长曲线。根据菌株生长曲线改变其摇瓶发酵的诱导温度、诱导时间、诱导剂IPTG的浓度及加入诱导剂IPTG的时间,通过SDS-PAGE分析hEDN表达情况,筛选最佳发酵条件。
1.2.3.1 诱导温度
挑取重组菌株的单菌落,接种于15 mL LB培养基中,37 ℃,200 r/min培养12 h,以1∶50体积比接种于TB培养基中,37 ℃,200 r/min培养至OD600=1.4~1.6时加入IPTG至终浓度为0.2 mmol/L,分成3组,分别于16、20、25 ℃继续培养36 h,其他条件保持一致,12 000 r/min,5 min收集菌体。
1.2.3.2 诱导时间
挑取重组菌株的单菌落,接种于15 mL LB培养基,37 ℃,200 r/min培养12 h,以1∶50体积比接种于TB培养基,37 ℃,200 r/min培养至OD600=1.4~1.6时加入IPTG至终浓度0.2 mmol/L,分成3组,分别于20 ℃继续培养12、24、36 h,其他条件保持一致,12 000 r/min,5 min收集菌体。
1.2.3.3 诱导剂IPTG的浓度
挑取重组菌株的单菌落,接种于15 mL LB培养基,37 ℃,200 r/min培养12 h,以1∶50体积比接种于TB培养基,37 ℃,200 r/min培养至OD600=1.4~1.6时加入IPTG,分成5组,每组加入IPTG分别至终浓度0.1、0.2、0.3、0.4、0.5 mmol/L,20 ℃继续培养36 h,其他条件保持一致。
1.2.3.4 加入诱导剂IPTG的时间
挑取重组菌株的单菌落,接种于15 mL LB培养基中,37 ℃,200 r/min培养12 h,以1∶50体积比接种于TB培养基,37 ℃,200 r/min培养,分成6组,每组加入诱导剂IPTG的时间分别为2、4、6、8、10、12 h,加入IPTG至终浓度为0.3 mmol/L,20 ℃继续培养36 h,其他条件保持一致。
1.2.4 人源内皮抑素蛋白的纯化
1.2.4.1 人源内皮抑素融合蛋白的纯化
用2 mL/min去离子水清洗PrePack GSH Purose 4 Fast Flow预装柱约20 min,去除乙醇;以2 mL/min流速、pH 8.0 PBS (140 mmol/L NaCl,2.7 mmol/L KCl,10 mmol/L Na2HPO4,1.8 mmol/L KH2PO4)平衡柱料约20 min;将过膜的hEDN融合蛋白上清液以1 mL/min的流速进样,以2 mL/min的流速用pH 8.0 PBS再次平衡柱料约20 min;然后以1 mL/min的流速用洗脱缓冲液(50 mmol/L Tris-HCl,10 mmol/L还原型谷胱甘肽,pH 8.0)进行洗脱约20 min,分别收集洗脱峰进行SDS-PAGE分析。另将得到的上述洗脱液进行超滤浓缩并且置换溶液,置换成50 mmol/L Tris-HCl溶液(pH 7.0),便于后期PreScission 蛋白酶(PreScission protease,PPase)酶切处理。
1.2.4.2 重组PPase切割融合蛋白及质谱鉴定
重组PPase是由human rhinovirus type 14 3C protease和GST组成的融合蛋白,该蛋白酶可在低温下特异识别短肽Leu-Glu-Val-Leu-Phe-Gln-Gly-Pro,并在Gln和Gly氨基酸残基之间进行酶切。
PPase酶活力定义(U):在5 ℃条件下反应16 h,能够切割10 μg的GST标签的融合蛋白达90%以上所需的酶量定义为1个活性单位。
将PPase以20 U酶活力的量加入到含有200 μg的hEDN融合蛋白中,反应体系包含50 mmol/L Tris-HCl,pH 7.0,150 mmol/L NaCl,1 mmol/L EDTA和1 mmol/L DTT的缓冲液,在5 ℃条件下进行酶切,16 h 和24 h后取样,SDS-PAGE检测酶切效果。
回收SDS-PAGE凝胶GST标签及hEDN条带置于1.5 mL EP管中,然后对凝胶进行脱色、孵育、冻干、消化、点样等预处理,用MALDI-TOF/TOF分析。
2 结果与分析
2.1 人源内皮抑素表达质粒的构建及验证
2.1.1 单基因表达质粒的构建及验证
按照1.2.1.1的方法成功构建了单基因表达质粒pET28a-hedn。酶切结果如图1-a所示。
2.1.2 融合标签共表达质粒的构建及验证
按照1.2.1.2的方法成功构建了融合标签共表达质粒pTIG-hedn、pETDuet-1-TrxA-hedn、pETDuet-1-hedn-TrxA、pETDuet-1-mbp-hedn、pETDuet-1-hedn-mbp。酶切结果如图1-b~图1-f所示。
2.1.3 融合表达质粒的构建及验证
按照1.2.1.3的方法成功构建了融合表达质粒pET32a-hedn、pGEX-6p-1-hedn。酶切结果如图1-g和图1-h所示。
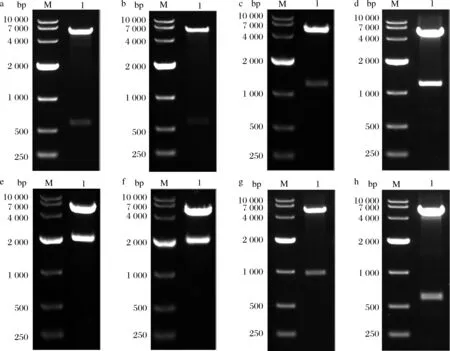
M-Marker,DL-10 000(Takara);1-重组质粒酶切结果a-pET28a-hedn;b-pTIG-hedn;c-pETDuet-1-Trx A-hedn;d-pETDuet-1-hedn-Trx A;e-pETDuet-1-mbp-hedn;f-pETDuet-1-hedn-mbp;g-pET32a-hedn;h-pGEX-6p-1-hedn图1 重组质粒酶切验证Fig.1 Restriction enzyme digestion analysis of recombinant plasmids
2.2 人源内皮抑素的诱导表达及质谱鉴定
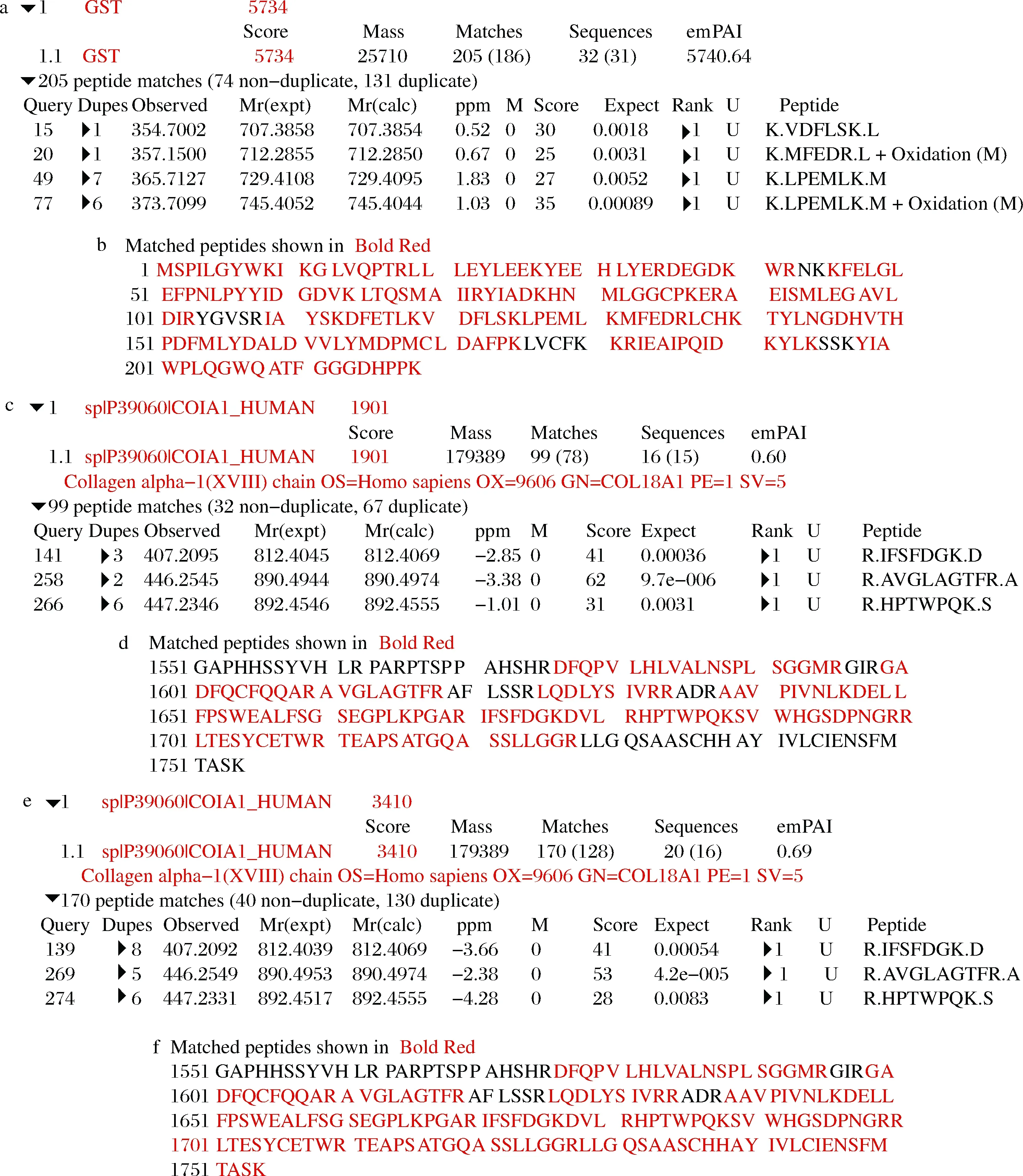
将上述含有hedn表达质粒的菌株及对应空载菌株进行摇瓶发酵,16 ℃,200 r/min,0.2 mmol/L IPTG诱导24 h,将破碎后的上清液及沉淀进行SDS-PAGE分析,结果如图2所示。本文构建的所有单基因表达菌株及融合标签共表达菌株发酵培养获得的重组蛋白经SDS-PAGE分析均未得到与hEDN分子质量理论值22 kDa相一致的条带,结果如图2-a~图2-f所示。融合表达菌株E.coliBL21(DE3)/pET32a-hedn理论上表达的融合蛋白大小应约为44 kDa,但是并未检测到相应大小的条带,如图2-g所示。融合表达菌株E.coliBL21(DE3)/pGEX-6p-1-hedn得到了与理论值相一致的融合蛋白,大小约为47 kDa,且对照组未检测到相应条带,如图2-h所示。根据上述结果,初步确定仅有E.coliBL21(DE3)/pGEX-6p-1-hedn能成功表达hEDN。进一步通过MALDI-TOF/TOF鉴定,结果如图3所示,破碎后上清液蛋白得分为128,破碎后沉淀蛋白得分为161,并且得分在质谱结果中排名靠前,表明结果可信,进一步确认hEDN的成功表达。
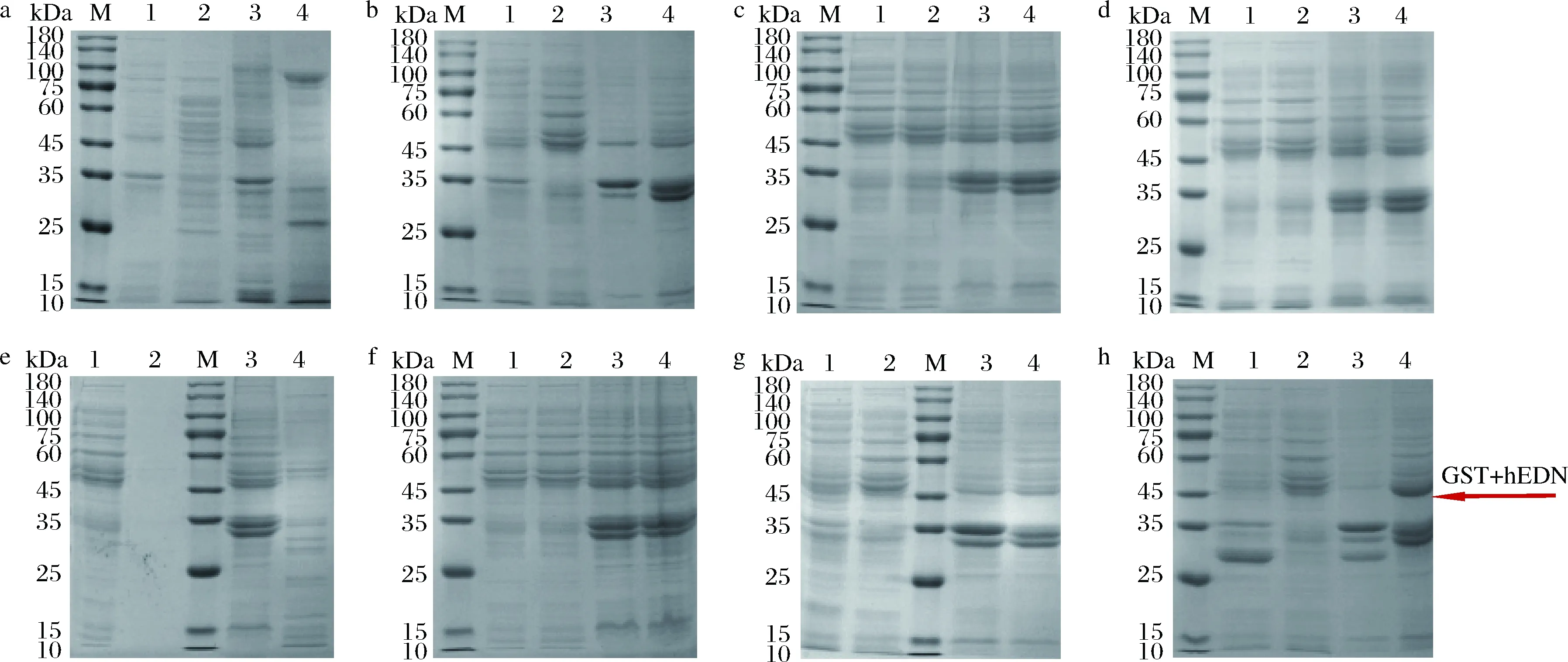
M-Marker;1-空载破碎后上清液;2-空载破碎后沉淀;3-重组菌株破碎后上清液;4-重组菌株破碎后沉淀a-E.coli BL21/pET28a-hedn;b-E.coli BL21/pTIG-hedn;c-E.coli BL21/pETDuet-1-Trx A-hedn;d-E.coli BL21/pETDuet-1-hedn-Trx A;e-E.coli BL21/pETDuet-1-mbp-hedn;f-E.coli BL21/pETDuet-1-hedn-mbp;g-E.coli BL21/pET32a-hedn;h-E.coli BL21/pGEX-6p-1-hedn图2 不同重组菌株的蛋白表达情况Fig.2 Protein expression of different recombinant strains
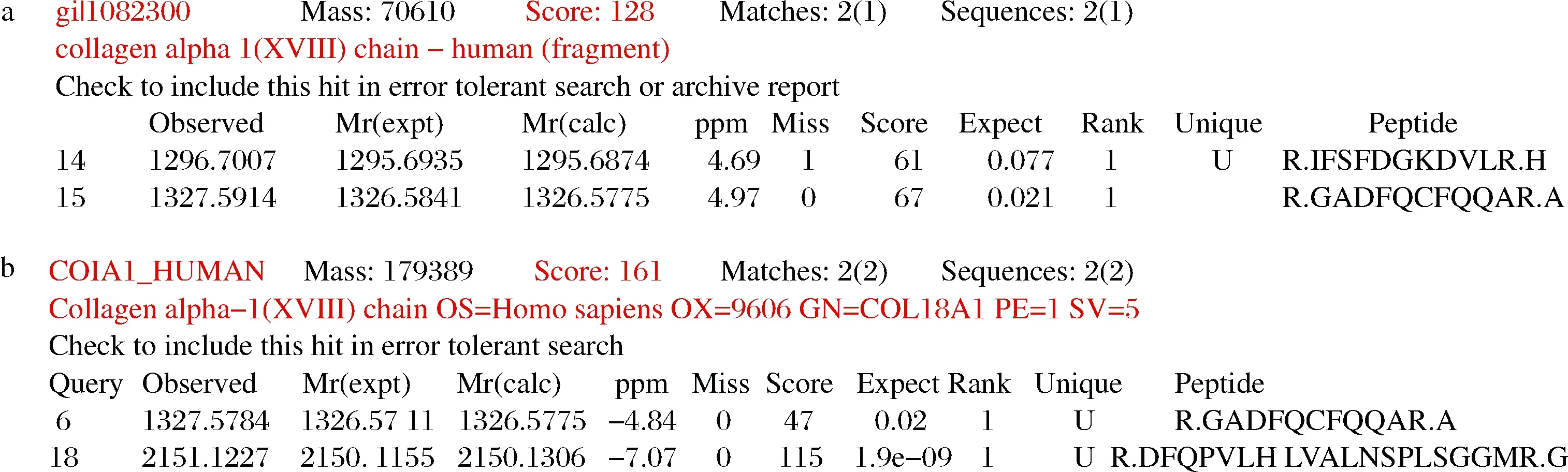
a-hEDN融合蛋白破碎后上清液;b-hEDN融合蛋白破碎后沉淀图3 hEDN融合蛋白的MALDI-TOF/TOF结果分析Fig.3 MALDI-TOF/TOF analysis of hEDN fusion protein
2.3 人源内皮抑素融合蛋白诱导条件的优化
E.coliBL21(DE3)/pGEX-6p-1-hedn菌株生长曲线如图4所示,该菌株在16 h左右基本进入稳定期。依据该菌株的生长曲线,分别对诱导温度、诱导时间、诱导剂IPTG浓度及加入诱导剂IPTG浓度条件进行发酵优化。
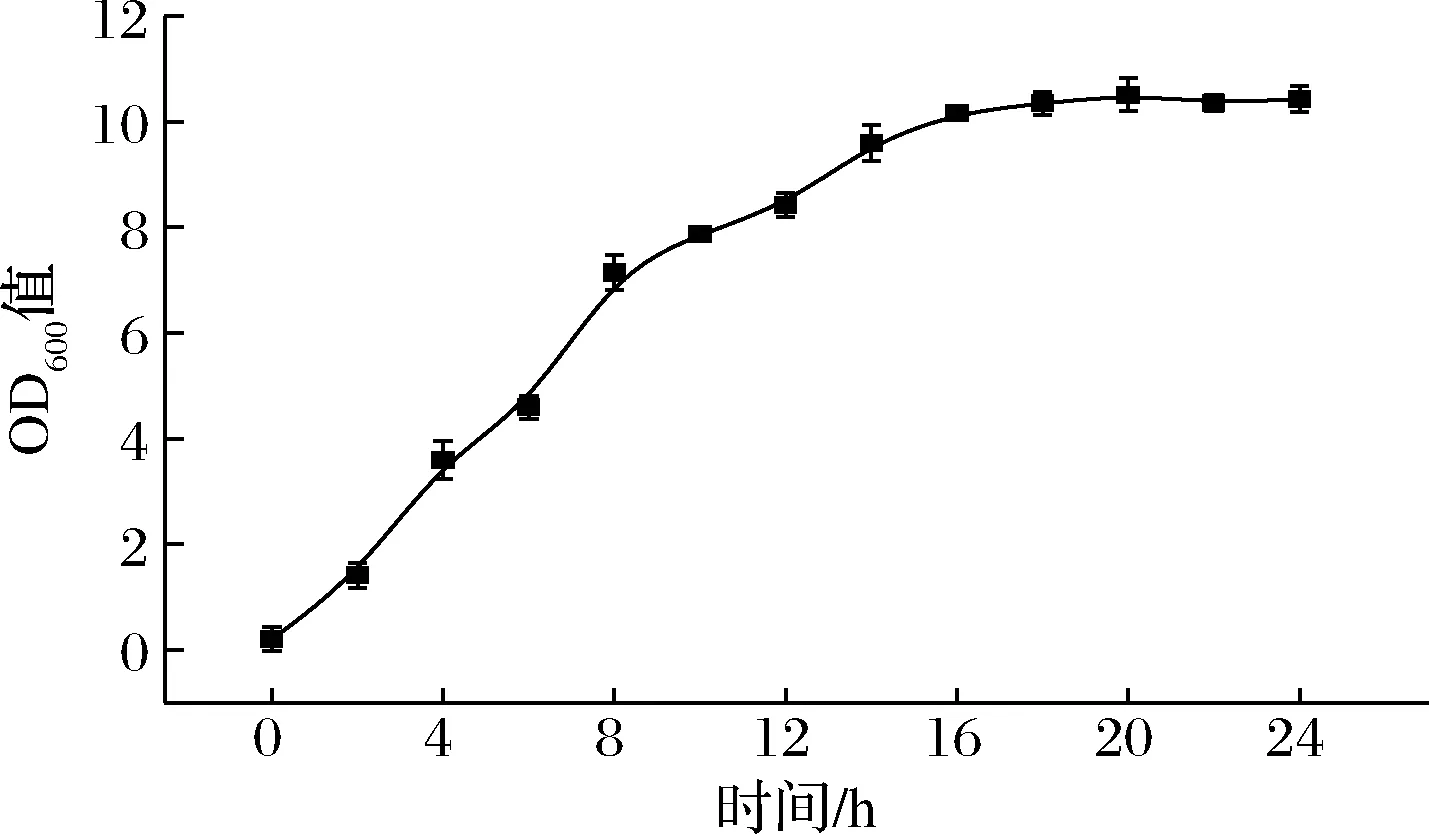
图4 重组菌株的生长曲线Fig.4 Growth curve of the recombinant strain
2.3.1 诱导温度
按照1.2.3.1方法,结果如图5-a和图5-b所示,细胞破碎上清液中明显16 ℃表达量最少,20 ℃和25 ℃无明显差别;但是这3个温度中25 ℃表达的包涵体明显是最多的,所以选择20 ℃作为最佳诱导温度。
2.3.2 诱导时间
按照1.2.3.2方法,结果如图5-c和图5-d所示,细胞破碎上清液中诱导12 h表达量最少,诱导36 h表达最多;并且诱导12 h包涵体最多,诱导36 h包涵体最少,因此选择诱导36 h 最为合适。
2.3.3 诱导剂IPTG浓度
按照1.2.3.3方法,结果如图5-e和图5-f所示,从不同浓度IPTG诱导的效果来看,hEDN融合蛋白包涵体的表达无明显差别,但细胞破碎上清液是有细微区别的,0.3 mmol/L的IPTG诱导效果稍好一些,因此选择0.3 mmol/L IPTG作为最佳诱导剂浓度。
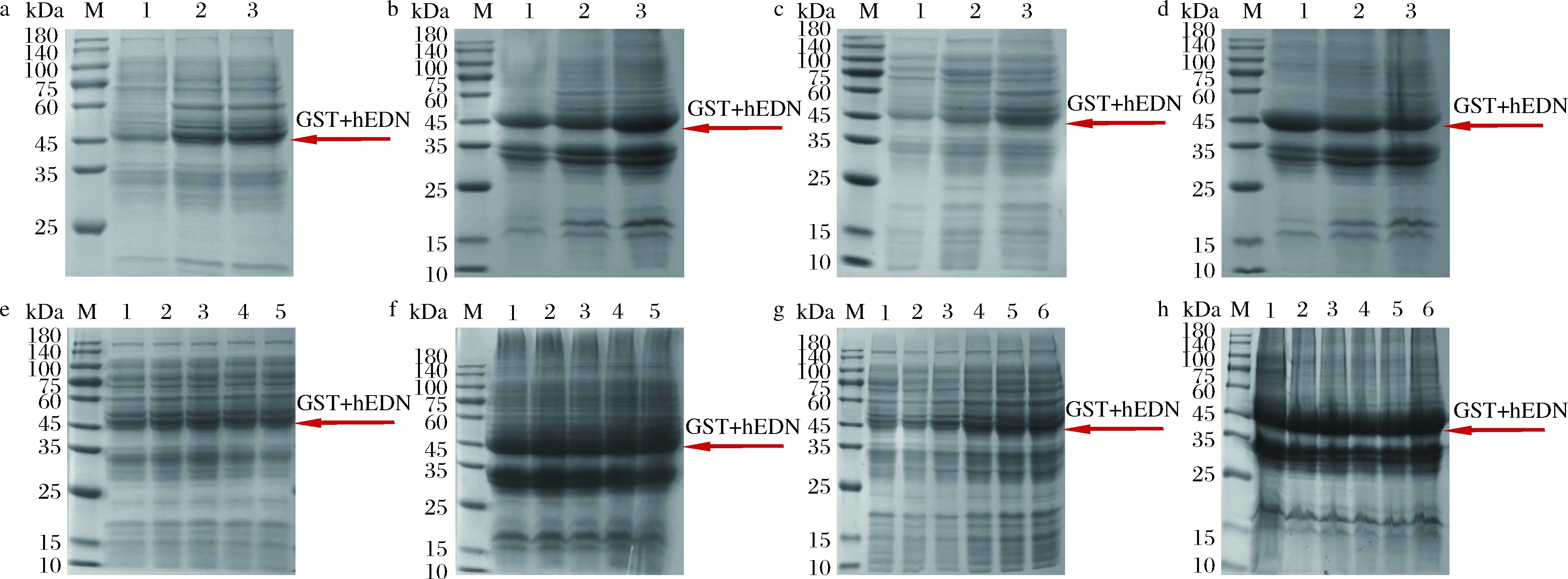
M-Marker;a,b-1-16 ℃,2-20 ℃,3-25 ℃,c,d-1-12 h,2-24 h,3-36 h,e,f-1-0.1 mmol/L,2-0.2 mmol/L,3-0.3 mmol/L,4-0.4 mmol/L,5-0.5 mmol/L,g,h-1-2 h,2-4 h,3-6 h,4-8 h,5-10 h,6-12 ha-诱导温度-破碎后上清液;b-诱导温度-破碎后沉淀;c-诱导时间-破碎后上清液;d-诱导时间-破碎后沉淀;e-诱导剂浓度-破碎后上清液;f-诱导剂浓度-破碎后沉淀;g-加入诱导剂时间-破碎后上清液;h-加入诱导剂时间-破碎后沉淀图5 发酵优化SDS-PAGE分析结果Fig.5 SDS-PAGE analysis results of fermentation optimization
2.3.4 加入诱导剂IPTG时间
按照1.2.3.4方法,结果如图5-g和图5-h所示,在2~12 h添加诱导剂时,hEDN融合蛋白包涵体的表达无明显差别,而8、10、12 h添加诱导剂时细胞破碎上清液中hEDN融合蛋白的表达效果优于2、4、6 h添加诱导剂,且在8、10、12 h添加诱导剂时上清液中hEDN融合蛋白的表达无明显差异。
2.4 人源内皮抑素蛋白纯化
2.4.1 人源内皮抑素融合蛋白纯化
因hEDN和GST标签融合表达,所以可溶性部分先过1次PrePack GSH Purose 4 Fast Flow预装柱初步纯化,SDS-PAGE分析显示在47 kDa处得到相应的理论值条带,表明初步纯化得到hEDN融合蛋白,但同时SDS-PAGE结果显示在约25 kDa处出现1条明显条带,飞行时间质谱鉴定为GST标签蛋白条带(图6-a、图6-c、图6-d)。
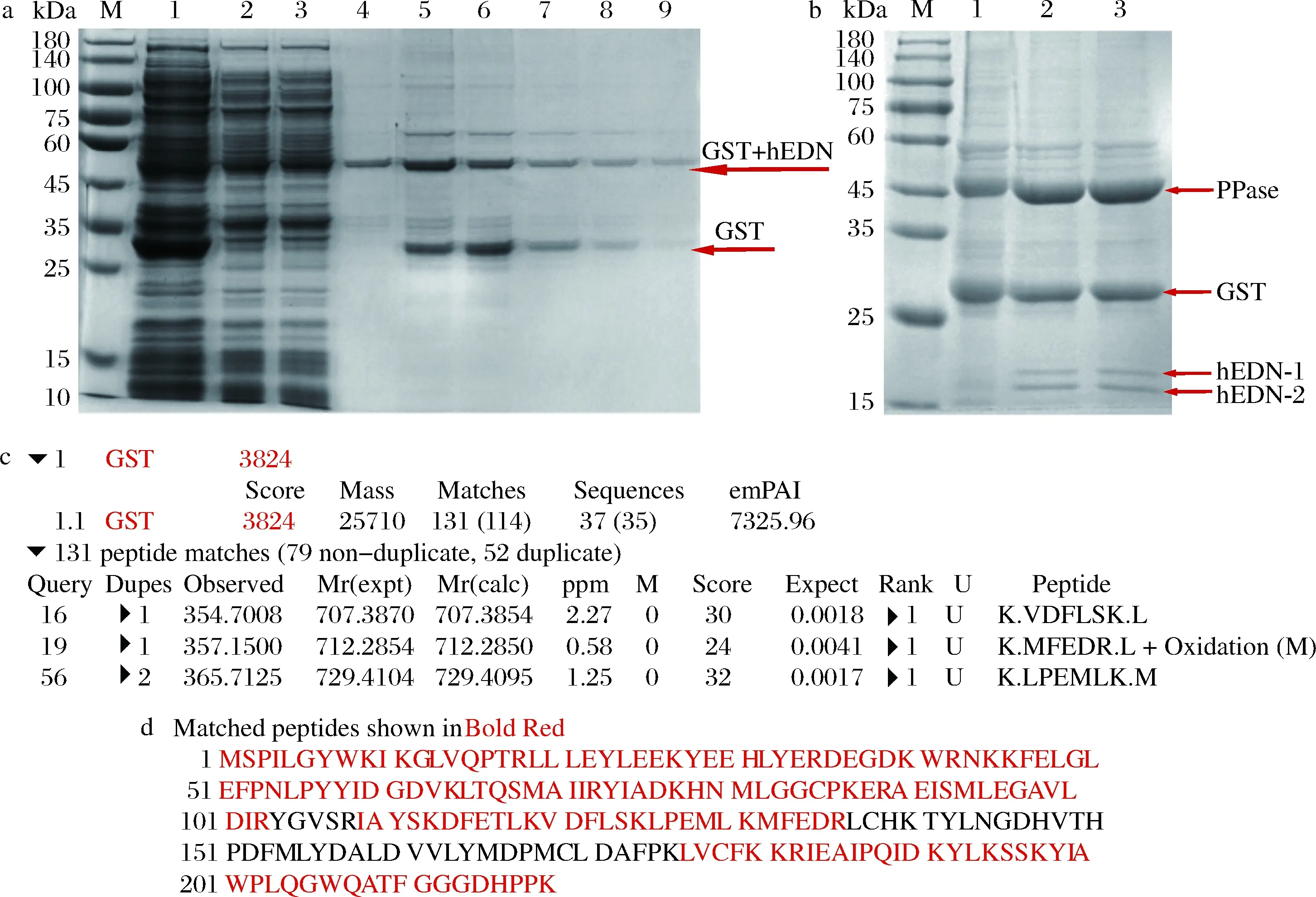
M-Marker;a-1-空载破碎后上清液;2-hEDN融合蛋白破碎后上清液;3-穿透液;4~9-收集液;b-1-收集液浓缩;2-酶切16 h;3-酶切24 ha-纯化后的hEDN融合蛋白的SDS-PAGE分析;b-hEDN融合蛋白酶切后的SDS-PAGE分析;c-鉴定GST标签结果;d-鉴定GST标签氨基酸序列结果图6 hEDN融合蛋白纯化及酶切后结果分析Fig.6 The results of the purified hEDN fusion protein and its enzymatic hydrolysate by the PPase
将纯化收集得到的样品进行超滤浓缩且置换成50 mmol/L Tris-HCl溶液(pH 7.0),为下一步切除GST标签做准备。
2.4.2 重组PPase切割融合蛋白
初步得到的纯化样品是包含hEDN和GST标签的融合蛋白,需要将GST标签切掉通过再次纯化得到hEDN。PPase是能够切割GST标签的蛋白酶,酶切16 h后经SDS-PAGE分析在25 kDa以下检测到两条条带(hEDN-1和hEDN-2),如图6-b所示,这两条带从上到下的质谱结果依次如图7所示,结果显示这两条带都是hEDN。
a-鉴定GST标签结果;b-鉴定GST标签氨基酸序列结果;c-鉴定hEDN-1结果;d-鉴定hEDN-1氨基酸序列结果;e-鉴定hEDN-2结果;f-鉴定hEDN-2氨基酸序列结果图7 hEDN融合蛋白酶切后的MALDI-TOF/TOF结果分析Fig.7 MALDI-TOF/TOF results of hEDN fusion protein after digestion by the PPase
3 讨论
大肠杆菌表达系统具有良好的遗传背景、低成本培养和高生产等优点,因而大肠杆菌表达系统是重组蛋白表达的首选[16]。然而,重组hEDN在大肠杆菌中容易形成包涵体。为了探究hEDN可溶性表达条件,本研究尝试在大肠杆菌中建立不同的表达体系,最终在所构建的重组菌株中仅有GST融合标签成功实现了hEDN的可溶性表达。值得注意的是,E.coliBL21(DE3)/pETDuet-1-mbp-hedn菌株转接至发酵培养基诱导表达后,发现培养基较澄清,表明该菌株并没有生长。hEDN较难在大肠杆菌中异源表达,可能是由于hEDN对细胞具有毒副作用,此外hEDN的表达依赖于胞内细胞器,而大肠杆菌中缺乏能促进蛋白成功表达的细胞器[24]。本文GST标签实现了hEDN的可溶性表达,并通过优化hEDN融合蛋白的发酵条件可知,重组菌处于对数生长后期时再加入诱导剂会增加hEDN融合蛋白的表达量。将细胞破碎上清液经过GST亲和层析初步得到hEDN融合蛋白,同时也得到了较浓的GST标签蛋白,可能是因为融合蛋白不稳定断裂或胞内蛋白酶切割造成的[25]。此外,hEDN融合蛋白经PPase酶切后理论上应得到hEDN,GST标签和PPase 3个部分,但是在SDS-PAGE分析中在15~25 kDa中得到两条条带,经过MALDI-TOF/TOF鉴定这两条带均为hEDN,这可能是由于PPase不正确切割所致,因为PPase对切割位点识别的敏感性受到核心区域两端氨基酸残基的影响[26],或者是由于不适宜的酶切条件引起二次切割所导致的[27],后续可对酶切条件进一步探索。
4 结论
本文利用GST融合标签表达策略实现了hEDN的可溶性表达,并通过发酵优化进一步确定了hEDN融合蛋白的最佳表达条件(8~12 h添加终浓度0.3 mmol/L IPTG,20 ℃诱导36 h)。该研究为人源多肽的可溶性表达与纯化提供了研究思路,同时对微生物发酵制备功能性多肽具有一定的借鉴意义。下一步,将通过细胞实验对hEDN功能进行验证。
