深度学习在生物序列分析领域的应用进展
2022-08-19张冀东王志晗
张冀东,王志晗,刘 博
(北京工业大学信息学部,北京 100124)
核酸与蛋白质是生物学研究中2个重要的研究领域.核酸是脱氧核糖核酸(deoxyribonucleic acid, DNA)和核糖核酸(ribonucleic acid, RNA)的总称.其中,具有遗传效应的DNA片段称为基因.基因与许多生物过程高度相关,在相关研究中,基因表达是研究基因的重要领域之一,它是指将来自基因的信息用于合成功能基因产物的过程[1-2].测序作为研究基因表达的重要手段,为直接观测DNA及RNA信息提供便利,加快了生物学和医学的研究与发现[3].在生物学与医学等研究领域中,获取DNA与RNA序列信息已成为必不可少的基础研究技术,包括诊断癌症在内的多种疾病[4],同时也可以用于表征抗体库[5-6],识别和分类更多的生物[5].RNA测序常用来分析不断变化的基因转录过程[7-8].基因表达的差异[9],除了信使RNA(mRNA)转录本,RNA测序还可查看不同的RNA群体[10].RNA测序的最新进展包括单细胞测序和固定组织的原位测序[11].现代高通量测序技术的大力发展,尤其是二代测序技术,使以核酸序列为基础的基因数据可以大规模产出,为生物信息分析提供了大量的数据与可靠的技术支持,也使得相关大数据的准确快速处理显得尤为重要.
蛋白质作为生物研究领域重要组成部分之一,随着生物技术不断的发展,其数据规模也不断增大.自1997年起,蛋白质研究领域中又分出一个重要的研究方向,即蛋白质组学.它是对蛋白质尤其是在结构和功能上的大规模性质的研究.蛋白质组会随着时间的推移及环境的不同而产生变化[12],使得基础的蛋白质组也需要被鉴定.在早期的蛋白质研究中,往往依靠RNA分析来完成相关研究[13-14],但是RNA并不是总跟蛋白质相关,已知的mRNA中也有不参与翻译蛋白质过程的.蛋白质组学的出现,为蛋白质直接度量提供了依据.蛋白质组学主要是从定性和定量2个方面对蛋白质进行研究.利用相关技术会产生大量的原生数据,经过复杂的处理与注释,便成为带有蛋白质信息的生物信息数据.
综上所述,生物序列数据主要包含核酸和蛋白质数据.这类数据往往具有数量大、维度高、相关性较为复杂等特点.传统生物学分析方法的综合分析能力较低,相关性较差,结果不准确,分析速度慢,对操作人员要求较高,往往需要富有经验的专家进行操作,因此,制约了现代生物学领域的研究发展.鉴于此,生物信息数据的快速处理及综合准确分析引起了广泛的关注.
近年来,基于大数据处理的另一研究领域——深度学习技术脱颖而出,作为交叉学科也逐渐应用在生物信息领域,并开始成为生物信息研究热点.2006年,深度学习概念首次出现在机器学习的研究中[15].它是基于深度神经网络的一系列机器学习技术[16],最早称为层次学习,在模式识别相关领域中有许多研究.不同于传统机器学习,深度学习更加关注从原始数据和低级数据中抽取特征,无需过多、复杂的数据特征预处理技术.深度学习可以利用多层次的结构,通过非线性处理抽取抽象特征结构.多层非线性处理指当前层将前一层的输出作为下一层的输入,在层之间建立起层次结构以判断数据的有用或无用.深度学习可以将复杂的数据转化为计算机可学习的表示方式,并从中抽取潜在的高维数据特征.这种抽取大型数据潜在隐含信息的能力是深度学习的主要优势.因此,近年来将深度学习技术应用于大数据量基因与蛋白质相关领域的研究得到了快速的发展,并取得了重要的研究成果,也成为目前2门学科的发展热点.本文就深度学习在生物大数据处理方面的优势和特点及其在基因与蛋白质领域中的重要研究成果进行梳理与阐述,并展望未来的发展前景.
1 主要的深度学习模型
伴随着图形计算等硬件的飞速发展,众多深度学习模型已应用于计算机视觉[17]、机器视觉[18]、语音识别[19]、自然语言处理、音频识别[20]、社交网络过滤和机器翻译等领域,甚至在某些特定场景下的表现超出人们的预期[21].这些证明了其强大的功能和广泛的适用性.当前主要应用于生物数据研究领域中的2种经典深度学习模型为循环神经网络(recurrent neural network, RNN)和卷积神经网络(convolutional neural network, CNN).除这2种主流神经网络模型外,近几年图卷积网络(graph convo-lutional network, GCN)也逐渐应用于蛋白质的相关分析之中.本文将以这3个神经网络模型作为重点进行介绍.
1.1 CNN
CNN已经广泛应用于生物信息数据的分析之中,如在染色体的可及性、DNA与RNA结合蛋白的结合位点预测以及甲基化等任务中都具有良好的表现.CNN首先由LeCun等[22]提出,其设计的初衷是为了处理网格化的数据,主要的数据对象为传统图像数据.CNN中比较重要的特征处理方法为卷积、激活和池化.经过卷积运算后的特征,会由激活函数进行激活操作,常见的CNN中激活函数多为非线性映射函数,而后模型会对激活后的特征进行池化操作.池化是一种降采样技术,可以降低特征图或者特征向量的尺寸.以图像为例,该方法会降低特征图的分辨率,过滤冗余细节并在提取潜在高维特征的同时,扩大感受野,捕捉更多的特征.一个卷积层中对输入数据的特征提取完成后,会输入到下一个卷积层对已经得到的特征图进行更进一步的高维特征捕捉,堆叠多个卷积层,逐步构建成一个完整的CNN.同时,通过反向传播,不断以自学习的方式更新滤波器中的特征权重,完成对输入数据的分析并根据现实任务做出相应预测.
1.2 RNN
另一种常见的深度学习模型是RNN.生物信息领域中,RNN往往是通过与CNN配套使用来发现CNN分析后的数据中远距离依赖等问题.RNN也有众多变体,如长短期记忆(long short-term memory, LSTM)模型[23]、门控循环单元(gated recurrent unit, GRU)等.RNN的出现是为处理序列数据的信息,尤其经常用在处理类似于语言文字等前后存在关联性的数据.RNN由输入层、隐藏层和输出层组成.相对于CNN等其他深度学习模型,RNN输入信息不只有当前输入值,还有上一次的输出值和其对应的权重矩阵,从而使得这一次的输出结果包含着上一层单元的处理信息.RNN本身也可以利用反向传播算法对权重矩阵进行更新.
1.3 GCN
GCN是近2年以来极为受人瞩目的一种图神经网络,逐渐被用于生物非欧氏数据分析之中,例如分析生物有机大分子物质的空间结构.早期GCN借鉴了RNN的方式进行节点信息的聚合[24-25]以提取数据特征,然而这种方式会造成网络开销过大、模型效能降低等问题.得益于CNN的成功表现,将卷积算法与图神经网络融合,发展成为GCN.与之对应的图卷积运算主要分为2类:谱图卷积和空域图卷积[26].谱图卷积是利用图傅里叶变换操作原始数据使之满足卷积定理,实现卷积操作[27],随后一些研究者又在此基础上进行了改进[28-29].但是,谱图卷积也存在着一些问题,例如只能处理无向图等.这使得对于图卷积的研究重新回到了空域研究上,如传播CNN[30]以及结合注意力机制[31]的图注意力网络[32].空域图卷积模型主要是参考传统CNN的方式在空域上对节点信息进行融合以实现对数据的分析.
2 深度学习在生物序列领域的研究现状
生物学领域利用测序、质谱等分析方法获取相关数据,经过清洗、标注后可以利用深度学习模型进行预测分析.利用深度学习分析数据的一般框架如图1所示.本文将从核酸与蛋白质领域对深度学习在生物信息技术中的应用研究现状进行分析.

图1 深度学习在生物学领域的应用
2.1 核酸领域的应用
在核酸领域的研究中,多是面向DNA和RNA测序结果的研究.DNA是组成基因的主要组成单位,而RNA是由DNA转录而来,这些序列都包含许多重要的生物信息.然而,利用传统生物学实验的方式,需要通过合成其对应蛋白质并检测其功能来推测原始DNA的作用,这使得基因分析与研究往往过分冗长且要消耗巨大的人力物力.DNA序列本身数据量也较为庞大且复杂.以人类全染色体组中的DNA数据为例[33],最多的有将近50万条数据,最少的也有11万条数据.数据中包含编码区与非编码区等功能区域,如果对这些区域进一步细分,将会产生更具体的DNA数据,如内含子区域、外显子区域等.RNA序列信息尽管相对DNA数据量较少,但数据量也相当可观.以ELAVL1蛋白质结合位点来说[34],就有约25万条数据.基于此,利用以数据驱动的深度学习模型分析预测基因序列的功能作用逐渐成为研究热点,也取得很多重要的研究成果.本文总结了一部分公开数据集,如表1所示.

表1 部分公开的核酸数据集
2015年,Alipanahi等[36]在专业学术期刊NatureBiotechnology上提出了DeepBind模型,该模型将深度CNN应用于分析预测DNA、RNA与蛋白质结合的研究之中.它利用16个滤波器的卷积层,在DNA或者RNA序列上以步长为1、大小为24的窗口进行滑动扫描的方式来进行特征分析.在确定DNA结合蛋白序列特异性方面,该项研究使用了DREAM5 TF-DNA Motif Recognition Challenge中的蛋白质微阵列(protein binding microarrays, PBM)数据进行测试.DeepBind得到了在所有算法中最好的测试结果,并且在对体内得到的实验数据分析预测中,DeepBind也得到了最高分数.在识别RNA蛋白质结合位点方面,用了不同生物的RNA蛋白质结合位点数据进行预测,也进行了将体外数据训练模型应用于体内数据预测的实验,DeepBind在2种实验中都得到了较好的结果.DeepBind也可用于分析基因变异的影响,并通过突变图谱实现可视化.在实验中,DeepBind能够预测启动子中是否发生了可能致病的单核苷酸变异.DeepBind模型的出现,提供了利用神经网络分析DNA、RNA与蛋白质结合位点的新思路.
2015年,Zhou等[37]提出了DeepSEA模型,该模型适用于对表观遗传学的研究,主要应用于转录因子、核酸酶超敏感位点和组蛋白修饰及异构体的研究.它对大量已获得的染色体修饰数据进行学习,经过不断的训练,捕获序列特征,进而预测序列上碱基的突变对染色体修饰功能造成的影响.该模型由3层CNN和2层全连接层构成,卷积层每层分别有320、480和960个卷积核.在对转录因子等数据集的分析上,曲线下面积(area under curve,AUC)达到了0.958,取得了良好的分析效果.最后,作者对模型进行扩展,对多种基因突变数据集进行分类,其效果均优于非深度学习的方法.
Pan等[38]设计了iDeep模型,该模型利用CNN分析RNA序列信息,同时利用深度信念网络来分析RNA的结构信息,最终将2类信息融合用以分析预测RNA的结合位点和基序.该模型主要是利用神经网络,同时分析了RNA序列与蛋白质的结合位点的序列信息与RNA的结构信息.2018年,iDeep模型进一步提升,iDeepS模型被提出[39].将iDeep中原有的深度信念网络舍弃,换为CNN.主体是由2个并行的CNN与1个RNN构成,2个CNN中,一个分析序列信息,另一个分析RNA对应的结构信息,融合2个卷积分支的输出,得到新的特征向量.该特征向量将会通过LSTM模型来分析其中远距离依赖关系后得到最终结果.iDeepS在与DeepBind模型比较中,多项数据取得了更好的分析效果.
韩国技术科学院Chung等[40]提出了利用多个卷积模型、多尺度滤波器的方式分析RNA结合蛋白的结合位点序列与结构信息的模型.该模型在AUC评价标准中取得了不错的表现.这些都展现了深度学习在DNA、RNA与蛋白质结合分析中的良好的分析预测能力.
2017年,DeepCpG的提出实现了利用深度学习对单细胞水平下DNA甲基化的预测[41].该模型利用Bi-GRU学习胞嘧啶和鸟嘌呤双核苷酸信息,并利用CNN学习DNA序列信息.将2个网络获取的特征进行拼接后再由2层全连接层获得最终结果.
完善发展治江思路,开展治江重大课题研究。面对严峻的治江形势和艰巨的工作任务,作为水利部派出的流域管理机构,长江委坚定不移地贯彻科学发展观和可持续发展治水思路,按照国务院批复的 《长江流域综合规划》(以下简称长流规)和长江水利发展战略确定的目标和任务,紧密结合流域实际,广泛深入调查研究,切实加强顶层设计,开展治江重大课题研究,着力强化信息化等基础工作,进一步完善和发展新时期治江思路,统筹推进长江治理开发与保护,为经济社会可持续发展提供了可靠保障。
在核酸其他领域中,深度学习也有着不错的表现,例如遗传变异分析.2018年,谷歌公司在NatureBiotechnology提出一种检测变异基因的深度学习模型DeepVariant[42],提供了利用CNN处理分析基因中突变部分的新的解决思路.该方法创新性地将图像识别技术与基因突变分析结合起来,取得了良好的效果.该模型示意图如图2所示.
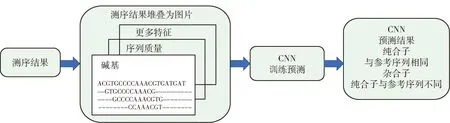
图2 DeepVariant模型的分析流程
在DeepVariant模型中,将测序结果与标准基因组作为对比,对比后的结果以红绿蓝图像保留.该模型为这种图像上3个通道的值重新做了定义,即红色通道为DNA的序列中的碱基,绿色通道为测序结果质量,蓝色通道为其他相关特征值.这些结果被分为三大类:与参考相同、杂合型、纯合变异.这些数据组成训练集与测试集,利用CNN进行分析预测.这样做的好处是巧妙地将对基因序列突变问题分析转变为对图像的分类操作,并且同时综合了碱基、测序质量值等指标.由于是将该问题转化为了图像的分类问题,所以可以利用图像领域中成熟的神经网络模型进行训练,如Inception v3[43]网络模型等.在与传统的变异检测方法比较中,DeepVariant在F1得分、召回率上都取得了良好的表现.
核酸领域另一个不可忽视的研究方向就是对DNA剪切位点的研究.DNA剪切位点是DNA打开双螺旋进行转录的初始位置,也是基因编辑的重要编辑位点.同时,检测DNA剪切位点也是反映DNA中基因变异的重要手段.当基因突变发生时,人体的DNA剪切位点相对于标准基因组的位置会发生改变甚至不表达.因此,快速查找出DNA中的剪切位点对于生物学核酸领域研究具有十分重要的意义.2019年,Jaganathan等[44]在Cell上提出了一个基于残差网络模型[45]的DNA剪切位点检测模型SpliceAI.在该模型中,考虑转录部分对检测查找剪切位点影响的同时将不参与转录的非编码区中的边翼序列也添加进训练集,从而将非编码区域对于编码区影响的关系特征也添加进模型之中.但是,目前这种非编码区的序列只考虑其存在的长度并不考虑其中具体的碱基序列.根据编码区两侧边翼序列的总体长度,将模型分成了4类,即SpliceAI-80nt、SpliceAI-400nt、SpliceAI-2k和SpliceAI-10k.在具体的模型实现中,SpliceAI参考了残差网络模型,设计了残差模块,由于数据维度高、体量大,放弃了传统池化层,利用空洞卷积扩大感受野.同时,为了更加具体地表现模型效果,利用了Top-k准确率作为评价标准.该评价标准是指特定某个指标的预测值与其对应真实值相同的数据量占该指标总体预测数据总量的百分比.最后,利用人类染色体数据对各个模型进行测试,其中表现最好的是SpliceAI-10k,Top-k准确率为0.95,最差的为SpliceAI-80nt,Top-k准确率为0.50.从上述数据结果来看,非编码区的边翼序列确实会对剪切位点甄别带来影响[44].
目前,在利用深度学习分析核酸序列的研究中,多是运用类比图片的方式,将DNA、RNA中4种不同的碱基对应卷积中的4种信道,再利用一维卷积进行分析.不同的研究区别在于分析预测的目标不同.在这些研究中,DeepVariant有效地将图像分析方式与DNA分析进行了结合,打开了对于核酸序列分析的新思路.
基于近年来的研究成果,本文总结了在基因领域应用较多的几种深度学习的常用模型,并就其模型基础以及应用目标进行了分类,具体的应用情况如表2所示.

表2 深度学习在核酸领域的应用
2.2 蛋白质领域的应用
深度学习也逐渐应用到了蛋白质的研究中.蛋白质的相关数据包括肽段质谱图、蛋白质有机高分子等数据集,而蛋白质有机高分子数据集又包含蛋白质互作网络数据集、蛋白质结构数据集(protein data bank,PDB)以及蛋白质对接基准数据集等.以PDB[46-47]和UniProt[48]为例,PDB中包含15万多个蛋白质结构数据,而UnitProt中共包含了约17万个蛋白质结构数据.本文对相关公开数据集进行了一定的汇总,如表3所示.与核酸研究类似,面对如此庞大的数据,作为数据导向的深度学习方法逐渐成为了研究热门.

表3 部分公开的蛋白质数据集
肽段的定量与定性分析是蛋白质组学中一项重要的研究内容,而这些分析又与其本身的质谱特征提取和理化性质密不可分.常见的分析方法为液相色谱分析,主要观测肽段色谱的保留时间.该保留时间一般是由其理化性质决定的,这往往需要对其理化性质进行进一步研究.因此,出现了很多推算其保留时间的算法,例如LUDE、GPTime等.但是,这些方法往往误差较大,对实际的推算造成了一定困难.鉴于这种情况,Ma等[49]提出了一种名为 DeepRT的模型.该模型基于CNN和RNN,从肽序列中自动学习特征,无须使用手工特征或设计规则.特征学习后,使用主成分分析(principal component analysis, PCA)方法进行降维,然后,使用3种常规的机器学习方法进行建模.该项研究使用2个已发布的数据集来评估DeepRT的性能,并且证明DeepRT的性能大大优于目前已知的几种常规方法.
液相色谱分析中对于特征提取的敏感度也是影响最终分析的重要因素.Zohora 等[50]提出的DeepIso由2个单独的基于深度学习的模块组成,可通过多层神经元来学习高维数据本身的多个表示形式,并适用于新获取的数据.该模型预测肽段的特征列表与基准数据集中对比实验结果达到了97.43%的高质量标识相匹配成功率.这些研究表明新颖的深度学习工具对蛋白质鉴定和定量方面的研究起到了一定的推进作用.
2020年,Alphafold在对蛋白质折叠的问题上取得了良好的效果[51].蛋白质作为维持生命所必需的生物大分子,其特有的空间结构往往决定了它的功能特性.例如,抗体蛋白通常为Y型,便于锁定病毒和细菌.但是,蛋白质的空间结构是极为复杂的,解析氨基酸残基形成的长链通过折叠形成的蛋白质结构,成为了一项重要的研究内容.Alphafold通过利用CNN,对氨基酸之间的距离以及其化学键的角度进行学习,给出一个推荐结构,再利用梯度下降算法调整优化整个结构预测结果.该方法利用深度学习方法,大大缩短了确定蛋白质结构的时间,在蛋白质研究方面具有重大的应用价值.
随着图卷积的迅猛发展,利用图卷积分析蛋白质的相关研究也逐渐增多.ProteinGCN[52]是一种基于图卷积方法对蛋白质空间结构进行分析的模型,模型示意图如图3所示.该模型能够在最新水平的蛋白质模型中实现较高的预测精度.此外,与之前提出的3D卷积网络相比,ProteinGCN中的自由参数数量少了1~2个数量级,不仅在蛋白质的空间结构上,而且在对蛋白质界面的分析中也会应用到图卷积.蛋白质界面预测在药物发现和设计中有重要的应用.Fout等[53]利用图卷积分析了蛋白质的三级结构,并将得到的信息用于分析界面残基之中,该方法显著优于常用预测手段.

图3 ProteinGCN模型分析流程图
在蛋白质的有关研究中,主要是利用CNN对蛋白质相关数据进行分析,以Alphafold为代表取得不错的效果.根据数据类型的不同,采用的CNN也略有不同.对于质谱图等图像分析,多采用二维CNN,而对于蛋白质序列的分析多采用一维CNN.新兴的GCN多用于研究蛋白质结构信息,随着技术的逐渐成熟,蛋白质未来发展将会逐渐向图卷积方向靠拢.
基于最近的研究成果,本文总结了在蛋白质领域应用较新的几种深度学习模型,并就其模型基础以及应用目标进行了分类,具体的应用情况如表4所示.

表4 深度学习在蛋白质领域中的应用
3 深度学习在生物序列分析中的应用局限性
无论是在核酸领域还是在蛋白质领域,利用深度学习对生物数据进行分析仍处于发展阶段.深度学习应用在生物学领域的最终目标是利用计算机强大的计算能力模拟复杂繁重的生物学实验过程,以期加快实验研究进展,但由于深度学习方法自身因素以及生物信息领域中的限制,距这一目标的实现还有较远的距离.现阶段,深度学习在生物信息分析中仍存在应用局限性,具体体现在以下几方面.
3.1 生物数据的复杂性
数据对于深度学习是最重要的实验基础,但生物数据具有高度的复杂性,使得深度学习在生物学中只能对单一目标进行分析.在基因数据中,描述一段基因,可以利用其序列信息、表达值信息、变异信息等,而这些信息之间可能又存在着相互关联.但是就目前深度学习分析而言,在综合分析生物数据方面还存在着欠缺,无论是对于DNA、RNA结合蛋白的分析,抑或是对于剪切位点的分析,都是对于基因中单一目标进行分析预测.在这方面,谷歌公司提出的DeepVarint[42]是一个良好的尝试,这种兼顾了基因序列与变异值的分析为将深度学习应用在基因分析之中提供了一个新思路、新方向.深度学习对于蛋白质分析的复杂性较高,主要是由于蛋白质本身信息体量较大,种类较多,例如大分子蛋白质结构的建模.
3.2 对生物数据预处理的依赖性
深度学习发展到今天,对优质数据的需求度越来越高,对数据预处理技术的依赖性也随之增大.生物数据具有高度的复杂性,使得对于相关数据的合理清洗与精确标注显得尤为重要.但是,这种依赖性在生物学上反而成了一定程度上的制约.生物数据不同于一般的图像、文字等数据,需要生物相关专业人员进行分析与标注,这就无疑增加了利用深度学习分析数据的成本,在一定程度上不利于轻量化生物学实验进程.尽管目前生物学数据有着一些公开数据库,例如NCBI、Esembel 与 UCSC等,但是这些公开数据的预处理仍需要良好的生物学领域知识.目前,这一问题的解决方案更多是依靠深度学习中的迁移学习将公开数据集的分析结果迁移到某一生物学领域之中,但已有模型参数是否可以合理迁移也成为相关研究人员重点关注的问题.解决这一问题就需要将深度学习领域与生物学领域交叉发展,进一步深化,联合制定更加具体的分析规则和程式化的分析方法.
3.3 深度学习可解释性的制约
深度学习本身的过程是一个“黑盒”的过程.在整个学习过程中,神经网络捕获的是人类难以理解的高维特征,这会使得研究人员对于神经网络分析结果的可信度产生一定的怀疑.对于机器自学习捕获的特征是否正确需要再利用其他技术进行验证评判.在图像分析任务中已经有不少方法可用来可视化机器分析过程,以判断机器是否捕获到合理的特征信息.生物数据相对于二维数据图像而言,序列化的核酸数据或者是三维的蛋白质结构数据难以通过直观的方法对机器的学习过程进行展示,使得生物研究人员往往需要利用传统生物学实验加以验证.随着深度学习在生物学领域的不断发展,已经将可视化技术应用在了基因领域,例如采用Saliency Maps[54]等技术可用于解释深度学习分析基因序列的过程.但特定任务的深度学习可解释方法并不普适,需要对不同任务设计相应的解释方案,实践难度较大.
4 总结与展望
深度学习相对于传统的机器学习,其优势在于可以利用复杂的多层次神经网络,依靠前向传播与反向传播,自行捕获学习数据的隐藏特征,并且利用梯度下降等算法对参数进行优化.这使得深度学习技术能够适应多类型、复杂、体量较大的数据,从而取得良好的预测效果.尽管深度学习在生物学领域中的应用仍处于起步阶段,但利用深度学习方法进行生物数据的分析与预测将是未来生物信息技术发展的重要方向之一.
近几年来,随着深度学习领域的不断发展,越来越多的新技术被应用到了核酸与蛋白质的分析之中.在核酸的研究领域中,目前应用最多的是CNN,无论是对染色体的可及性分析或者是对基因中存在的遗传变异分析均有良好的表现.但是,生物数据的复杂性和深度学习方法对数据预处理的高度依赖性同样是不可忽略的.因此,应用深度学习方法对未来生物数据的分析应有以下几点的考虑:
1)考虑算法的可迁移性.生物过程产生的数据复杂繁多,但是这些数据之间也存在着相关性.例如,在对蛋白质结构分析时,就应考虑是否可以利用同族蛋白质分析的深度学习模型进行迁移分析,加快分析进程.
2)加强2类领域交叉领域的发展.当前,生物数据的预处理对于深度学习研究人员仍是一大难题.因此,应深化交叉领域合作,制定一套程式化的分析方式,保证准确性的同时,缩短数据预处理的时间.生物研究人员也应对深度学习数据分析过程进行简要了解,辅助生物科学的研究
3)加强新技术的应用.纵观全文,可以发现,大部分的生物数据分析研究依然停留在一维CNN.但是,在深度学习领域,也有更多的新模型不断出现,应及时跟进最新技术,发现新的研究热点.
相信随着深度学习技术与以生物序列分析为代表的生物信息技术之间融合程度的不断加深,深度学习也将会成为生物数据分析的一个重要工具.
