临床症状轻微的TK2相关的线粒体DNA耗竭综合征1例
2022-08-09蒋姣,郭虎,金波
蒋 姣,郭 虎,金 波
南京医科大学附属儿童医院神经内科,江苏 南京 210008
线粒体DNA 耗竭综合征(mitochondrial DNA depletion syndromes,MDS)是由核基因突变引起线粒体DNA 合成或复制障碍的一类常染色体隐性遗传病,是儿童常见的呼吸链缺陷病,可导致新生儿和婴儿早期死亡。目前研究发现可分为4 型:肌病型、脑肌病型、肝脑病型、神经胃肠病型。其中肌病型致病基因是TK2基因和SLC25A4基因。TK2基因变异有多种临床表型,包括婴儿型、儿童型及晚发型。本文首先在国内报道1例TK2基因变异致临床症状轻微的MDS病型患儿的临床资料,并进行文献复习,以期提高临床医生对TK2基因变异所致的肌病型MDS临床及基因诊断的认识。
1 临床资料
患儿,男,12岁,因周围性面神经麻痹于外院住院治疗期间查血生化发现肌酸激酶(creatinekinase,CK)升高至1 464 U/L。体格检查:神志清楚,反应可,面色正常,面容对称,双瞳孔等大等圆,直径3 mm,对光反射灵活,眼球各方向活动正常,无眼睑下垂,心肺腹无异常,四肢肌力5 级,肌张力正常,膝腱反射正常引出,巴氏征阴性,指鼻试验、闭目难立征阴性,Gower 征阴性。患儿系G1P1 足月顺产,既往生长发育同一般同龄儿,父母体健,非近亲婚配,无同胞兄弟姐妹,否认家族性遗传病史。20 d 后复查肌酸激酶4 153 U/L;心电图检查示:窦性心律不齐;肌电图(EMG)检查示:所检神经感觉及运动传导速度均正常,所检肌肉无典型自发电位(纤颤电位及正尖波),无肌强直电位,轻收缩平均运动单位电位(MUP)时限缩短,波幅未见减低,多相波明显增多,重收缩时募集为混合⁃干扰相。结论:异常EMG(肌源性损伤改变);双小腿肌肉MRI提示:双小腿中上段肌肉轻度萎缩伴脂肪浸润(图1)。在外院行DMD基因检测未检测到DMD基因大片段缺失/重复。未能明确病因来我院就诊,追问病史,发现4年前开始长距离行走容易疲劳、体育课成绩差、不能跑着上楼梯,平素一般日常活动无受限。行安静时血气分析:pH 7.363,乳酸(Lac)1.1 mmol/L,碱剩余(BE)-2.7 mmol/L;平地快走蹦跳(中间反复休息多次)15 min 后血气分析:pH 7.349,Lac 6.6 mmol/L,BE-6.2 mmol/L。经家长知情同意后,采集患儿及其父母外周血标本各2 mL,送至北京迈基诺医学检验实验室进行神经肌肉病Pannel 检测,对阳性结果采用Sanger测序法进行位点验证及家系验证。检测到患儿TK2 基因c.547C>T、c.341A>T 复合杂合变异,Sanger家系测序提示c.547C>T变异来源于母亲,生物学致病等级ACMG 指南评定为可能致病:PS1+PM2+PP3,c.341A>T 变异来源于父亲,生物学致病等级ACMG指南评定为可能致病:PM1+PM2+PM3+PP3,该患儿突变位点生物信息学蛋白功能预测软件SIFT、PolyPhen_2、MutationTaster、GERP++、REV⁃EL 预测结果都分别为有害、有害、有害、有害、有害。其中c.341A>T 变异仅在中文文献中有报道。结合患儿临床症状及辅助检查,最终诊断为TK2基因相关的MDS(肌病型、儿童型),给予二磷酸果糖、左卡尼汀、复合维生素B、辅酶Q10治疗。
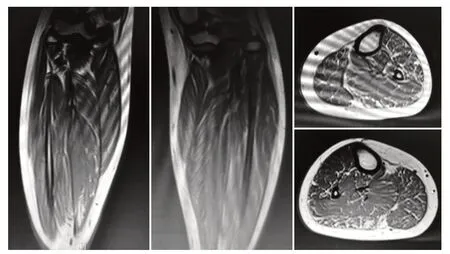
图1 患儿双小腿肌肉核磁共振
2 讨论
以“线粒体DNA 耗竭综合征”、“mitochondrial DNA depletion syndrome”、“TK2”、“thymidine kinase 2⁃deficient myopathy”等关键词,对中国期刊全文数据库(CNKI)、万方数据知识服务平台和美国国家生物技术中心(NCBI)、生物医学文献数据库(PubMed)数据库2001—2021 年6 月收录的文献进行检索,检索到中英文文献共157例TK2相关MDS患者,包含73种变异(图2),其中中国儿童共8例[1-5],均为2岁前发病,且其中7例存活年龄不超过2年。但本文报道患儿发病年龄晚,症状轻微。

图2 TK2相关MDS(肌病型)的73种变异
TK2 相关线粒体DNA 耗竭性肌病或者TK2 相关MDS(肌病型),是一种罕见的常染色体隐性遗传性疾病,其特征性的临床表现为婴幼儿期起病的进行性肌肉无力,伴或不伴肝脏、心肌以及脑组织等其他高能量代谢需求脏器受累。Garone 等[6]根据发病年龄将TK2相关MDS分为:婴儿型(≤1岁),儿童型(1~12 岁),晚发型(≥12 岁)。也有将2 岁内发病,并且未获得行走能力、需要机械通气或者发病1年内两者都有的定义为早发型[7],2~18 岁发病为青少年/儿童型,18岁以上为成人型[8]。TK2相关MDS主要累及肌肉,还有少数患者表现出非骨骼肌受累,包括听力损失、认知能力下降、脑病、QT 间期延长、心律失常、多发性骨折、肾小管病和男性乳房发育症。首发症状多表现为肌无力,运动里程碑延迟或运动不耐受,也可表现为呼吸功能不全、上睑下垂、复发性横纹肌溶解[9]等。Sara 等[10]通过尸检证实,TK2 相关MDS 患者除骨骼肌外,隔膜中线粒体DNA(mtDNA)含量特别低,而TK2 相关MDS患者的死亡常常是因为重症感染合并呼吸功能不全造成的。
研究表明,TK2 相关MDS 患者的CK 水平常升高但不具有特异性。在一项18 例晚发型患者的回顾性研究发现,94.4%的患者CK水平出范围升高至190~2 435 UI/L(正常水平<170 UI/L),16.7% 的患者显示水平比正常上限高出10 倍[11]。12例在基础条件下测血清乳酸水平,只有3 例(25%)显示水平略有增加(比正常上限高出1.4~2.0 倍)。但血清GDF⁃15水平升高,已被证实在线粒体肌病中具有诊断意义[12],且GDF⁃15 水平可能是评价治疗效果的潜在标志物[13]。EMG 检查提示以MUP 时限缩短为主要表现的肌源性改变。MRI 主要表现为脂肪浸润;肌肉病理改变亦无明显特异性,与其他线粒体肌病表现类似,包括肌纤维萎缩和/或坏死(28/36,69.5%)、纤维大小变异/1 型优势(24/36,66.6%)、中央核增加(11/36,30.5%)、肌浆空泡(5/36,13.9%),肌肉组织的脂肪替代(7/36,19.5%),以及结缔组织的纤维化或增加(14/36,38.9%)[6]。已研究的大多数患者的肌肉组织化学异常,包括细胞色素c 氧化酶(COX)染色肌纤维不着色和大量的参差不齐的破碎红纤维(raggedredfiber,RRF)[5]。早发型患者常常生存期不超过1 年,而儿童期或成人期发病的患者常常进展缓慢,生存期常超过10 年。本文报道患儿在8岁时出现症状,主要表现为运动不耐受,12岁确诊,对症治疗后CK 水平最低降至304 U/L,但有波动,截至确诊后1.5 年随访,患儿运动不耐受并没有得到改善,但进展缓慢,目前日常活动仍无明显受限。血清CK 水平升高明显,平静状态下Lac无升高,但活动后升高,EMG 提示肌源性改变,肌肉MRI 可及肌肉萎缩及脂肪浸润,与既往文献报道一致。
TK2相关MDS 是常染色体隐形遗传病,是编码胸苷激酶2(thymidine kinase 2,TK2)的核基因TK2突变引起的,TK2是一种核编码的线粒体酶,可催化脱氧胞苷和核苷胸苷生成核苷单磷酸酯,核苷单磷酸酯进一步磷酸化生成脱氧核苷三磷酸,参与到mtDNA 的复制中。TK2 致病性变异导致TK2 功能障碍,导致mtDNA 前体合成受损,从而引起mtDNA耗竭。自2001 年Saada 等[14]首次报道以来,目前已报道73种变异,在TK2的所有外显子中均发现了致病性变异,主要为错义突变,其他还包括框移突变、无义突变、剪切突变及大片段缺失。基于基因变异位点的定位,外显子5 为热点变异位置,而外显子8为TK2 突变的第2 个常见位置。外显子5 编码α4螺旋,是脱氧核苷激酶域中酶二聚化和核苷识别所必需的;外显子8 编码α8 螺旋,是结合ATP 的磷酸基团和催化作用所需的盖子区域的组成部分。我们报道的这例患儿为c.341A>T(p.Gln114Leu)、c.547C>T(p.Arg183Trp)复合杂合突变,突变位点分别位于外显子5 和8。目前报道的迟发型患者的基因型以p.Lys202del 或p.Thr108Met 纯合突变为主,均为西班牙人或西班牙后裔[11],但这2个热点突变位点同时也在早发型病例的复合杂合变异中发现[7],但该类患者存活时间均较长,所以这2 个位点的突变可能具有种族聚集性,表型相对较轻,但与发病年龄并没有一定相关性。同样p.Arg130Trp 变异在芬兰患者中发现[15]。而所有目前报道的临床症状符合的中国患儿均为复合杂合突变,并且其中大部分变异位点,包括c.497A>T(p.Asp166Val)、c.328C>Tp.Gln110*)、c.659T>C(p.Leu220Pro)、c.161G>A(p.Cys54Tyr)、c.557C>G(p.Pro186Arg)、c.341A>T(p.Gln114Leu)、c.619⁃2A>T、c.248C>T(p.T83M)、c.673C >T(p.Arg225Trp)、c.923A>G(p.Ter308Trp)在国际上均未被报道过,而c.341A>T(p.Gln114Leu)与本文报道患儿其中一个突变位点一致。无独有偶,俄罗斯报道的8例TK2相关MDS患者共发现7种突变位点,其中4 种未被报道过[16]。因此可以推测,TK2相关MDS在不同种族中突变位置可能不同。
TK2相关MDS 既往并无特效治疗方案,只能对症治疗,包括维生素、呼吸底物及辅酶等,但已有研究证实脱氧胞苷单磷酸疗法对TK2 缺乏的小鼠模型有效[17]。Cristina等[7]和Ana等[18]分别在16例和6例患者的临床试验中证实了脱氧核苷单磷酸和脱氧胞苷单磷酸盐疗法的安全性和有效性。随着医学的发展,遗传性神经肌肉疾病逐渐出现新的革命性治疗方法。诺西那生于2016 年被批准用于脊髓性肌萎缩的治疗,Ataluren 也成为第一个获批用于杜氏肌营养不良的治疗药物[19],TK2相关MDS也将可能有新的治疗方法。因此早期诊断并预防重症感染的发生,有利于提高生存率;并可能可以获得潜在的治疗方法从而提高生活质量。
综上所述,肌无力是TK2相关MDS肌病型主要临床表现,而呼吸衰竭是其主要潜在死因,TK2相关MDS肌病型具有基因型和表型异质性,早期基因检测有利于治疗和遗传咨询。
