薯蓣皂苷元无定形固体分散体制备及体内外评价
2022-07-21常金花薛禾菲王雨欣刘翠哲周剑宇
刘 沛,常金花,康 凯,薛禾菲,王雨欣,徐 林,刘翠哲,周剑宇
薯蓣皂苷元无定形固体分散体制备及体内外评价
刘 沛,常金花,康 凯,薛禾菲,王雨欣,徐 林,刘翠哲*,周剑宇*
承德医学院 河北省神经损伤与修复重点实验室,河北省中药研究与开发重点实验室,河北 承德 067000
制备薯蓣皂苷元无定形固体分散体(diosgenin amorphous solid dispersion,Dio-ASD),提高Dio溶出度和生物利用度。应用分子模拟技术分析Dio与载体之间相互作用并通过抑晶实验验证,构建Dio与载体混溶性曲线相图,理论预测二者混溶性;以Soluplus为载体,应用共沉淀法制备Dio-ASD;通过溶出度测定、差示扫描量热分析(differential scanning calorimetry,DSC)、X-射线粉末衍射(X-ray powder diffraction,XRPD)、扫描电镜分析(scanning electron microscopy,SEM)、傅里叶红外光谱(Fourier transform infrared spectroscopy,FT-IR)对Dio-ASD进行体外评价;采用UPLC-MS/MS方法测定大鼠体内Dio血药浓度,计算药动学参数,对Dio-ASD进行体内评价。分子模拟结果显示Soluplus与Dio之间能形成疏水键和氢键相互作用,结合能强于其他载体,且Soluplus对Dio的抑晶作用最强。构建了混溶性曲线相图,Dio与Soluplus在25 ℃下的混溶性为68.57%。与原料药相比,Dio-ASD溶出度明显提高。物相表征结果显示,Dio以无定形态存在于Dio-ASD中,Dio和Soluplus之间存在相互作用。药动学结果表明大鼠ig给药后,Dio-ASD的生物利用度较Dio提高了近5倍。制备的Dio-ASD可以显著提高药物溶出度和大鼠体内生物利用度。
薯蓣皂苷元;无定形固体分散体;分子模拟;混溶性相图;药动学
薯蓣皂苷元(diosgenin,Dio)俗称皂素,广泛存在于豆科和薯蓣科植物中[1],是一种植物自然合成的甾体皂苷元,属螺甾烷醇糖苷元。现代研究证实Dio具有抗癌、免疫调节、心血管保护、降血脂等多种药理作用[2]。然而,Dio具有强疏水性(lg5.7)和低水溶性(6.15 ng/mL),导致Dio的生物利用度较低[3]。为了提高其溶解性和生物利用度,目前有使用前体药物、纳米以及环糊精包合技术提高Dio口服生物利用度的报道[4-6],但已有策略或工艺复杂或效果不佳,Dio衍生物的制备需要多步合成操作,工艺较复杂;制备Dio环糊精包合物需要振摇24 h后干燥,制备时间较长;采用纳米混悬技术仅提高Dio生物利用度约2倍,效果不佳。因此,有必要开发一种简单有效的方法来提高Dio的生物利用度。最近,研究集中在无定形固体分散体(amorphous solid dispersion,ASD)上。ASD中活性药物成分以无定形态分散于载体材料中,表面自由能大,其粒径减小,比表面积大,进而提高溶解度和溶出度[7]。且ASD是一种典型的过饱和给药系统,可显著提高游离药物浓度,远远超过其在胃肠道中的平衡溶解度[8],通过浓度梯度赋予的驱动力,ASD可促进药物被动转运通过肠膜,从而增加药物的吸收[9]。本研究应用分子模拟技术分析Dio与载体之间相互作用并通过抑晶实验验证,构建Dio与载体混溶性曲线相图,理论预测二者混溶性,制备薯蓣皂苷元无定形固体分散体(diosgenin amorphous solid dispersion,Dio-ASD),对其溶出行为和物相表征进行考察,最后对其进行药动学和稳定性研究。
1 仪器与材料
1.1 仪器
Agilent 1260型高效液相色谱仪,美国安捷伦公司;Waters Acquity UPLC H-Class型超高效液相色谱仪,沃特世科技有限公司;Sciex Triple Quad 5500型系统质谱仪,AB Sciex公司;RC806型溶出试验仪,天津市天大天发科技有限公司;Sybyl 6.9.1软件,美国Tripos公司;AutoDock 4.0软件,美国Molecular Graphics Laboratory研究所;Materials Studio软件,美国Accelrys公司;TA DSC-250型差示扫描量热分析仪,美国TA仪器/沃特世科技(上海)有限公司;Bruker Tensor 27型傅里叶变换红外光谱仪,德国Bruker光谱仪器公司;Jeol-JSM-7500F型扫描电子显微镜(SEM),日本电子株式会社;PW3040/60 X-射线粉末衍射仪,荷兰帕纳科公司。
1.2 材料
Dio对照品,批号111539-200001,中国食品药品检定研究院;Dio原料药,批号84414368E0,南京春秋生物工程有限公司;聚乙烯已内酰胺-聚醋酸乙烯酯-聚乙二醇接枝共聚物(Soluplus)、羟丙基甲基纤维素E30(hydroxy propyl methyl cellulos E30,HPMC-E30)、聚乙烯基-吡咯烷酮K30(polyvinylpyrrolidone K30,PVP-K30)、普兰尼克(Pluronic-F68)、聚乙二醇4000(polyethylene glycol 4000,PEG4000),巴斯夫股份公司;十二烷基硫酸钠(sodium dodecyl sulfate,SDS),天津市福晨化学试剂厂;乙腈、甲醇,色谱级,北京迈瑞达科技有限公司;乙腈、甲醇,质谱级,赛默飞公司;娃哈哈纯净水,娃哈哈纯净水有限公司;屈臣氏蒸馏水,屈臣氏集团;其他试剂均为分析纯。
1.3 动物
健康雄性SD大鼠购自北京维通利华实验动物技术有限公司,许可证号为SCXK2016-0006。所有动物均暴露于温度为(22±1)℃,相对湿度为 (50±1)%,光/暗周期为12 h/12 h的环境中。大鼠在实验前自由食用标准鼠粮和无菌水,禁食12 h。所有动物相关方案均经承德医学院动物伦理实验委员会批准(批准号:NO.CDMULAC-20180410015)并符合《国家实验动物使用法》的要求。
2 方法与结果
2.1 Dio-ASDs载体的选择
2.1.1 分子模拟技术对Dio-ASD载体的选择 首先,利用Sybyl 6.9.1软件的Sketch Molecule模块构建载体的初始三维结构;其次,通过Minimize模块,采用Powell能量梯度法,选取AMBER7 FF99力场和Gasteiger-Hucel电荷,能量梯度为20.93 J/mol,收敛梯度为50 J/mol,最大迭代次数为10 000,对候选载体进行几何优化和分子力学优化,获得所有分子能量最低时的优势构象。采用AutoDock 4.0软件包进行分子对接,AutoDock 4.0具有AutoGrid和AutoDock 2个程序。首先,采用AutoGrid程序计算化合物中各种类型原子的格点能量;其次,采用AutoDock程序以药物分子为配体,候选载体分别为受体进行半柔性对接,在进行分子对接过程中,配体保持柔性、受体则保持刚性状态;参数设置如下:最大迭代次数为15 000,最大运算步数为3000,运算次数为100,最大群体大小为300,栏格分辨率为0.2 nm;最后,根据配体的不同构象、方向、位置及能量对各种结合模式进行评分并排序。结果见图1。
根据Dio与载体分子间相互作用及界面氢键分布,氢键(绿色虚线)和疏水作用(粉色虚线)是Dio和Soluplus之间形成的主要作用。Dio与HPMC- E30之间只形成氢键,不形成疏水作用,导致其结合能低于Soluplus。与HPMC-E30和Soluplus相比,Dio与PVP-K30、Pluronic-F68、PEG4000之间没有氢键和疏水作用,导致它们的结合能较低。计算出的结合能值Soluplus(−28 550 J/mol)>HPMC- E30(−27 503 J/mol)>PVP-K30(−27 334 J/mol)>Plur-onic-F68(−24 034 J/mol)>PEG4000(−23 493 J/mol)。因此,虽然Soluplus可以与Dio形成较强的相互作用,但从结合能值来看效果并不显著,下一步将通过实验进行验证。
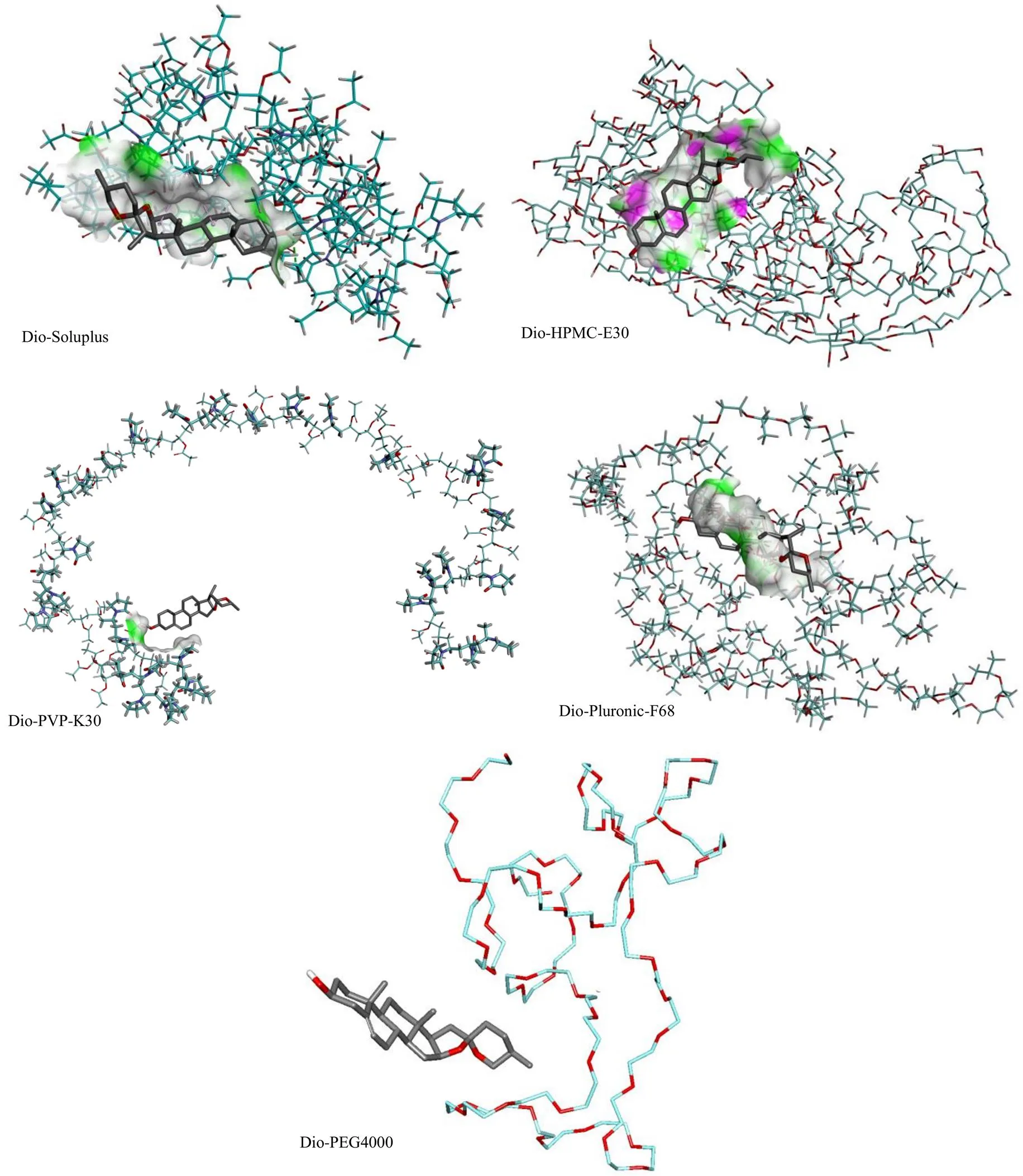
图1 Dio与不同载体分子对接示意图
2.1.2 不同聚合物对Dio超饱和溶液结晶抑制作用的考察 精密称取27 mg Dio溶于最少量的无水乙醇中,超声溶解,使成均一待结晶的超饱和溶液,用以模拟超饱和药物体系。精密称取162 mg聚合物溶于900 mL水中,平衡一段时间,将上述超饱和药物溶液迅速加入聚合物溶液中。以不加聚合物的溶液为对照,介质保持在37 ℃下,转速为50 r/min,分别于5、10、15、20、30、60、90、120、150、180、210、240 min时取样5 mL,并迅速补液5 mL,过0.45 µm微孔滤膜,弃去初滤液,取续滤液进行HPLC测定,实验重复3次。为避免因温度降低引起的任何药物结晶发生,取样的溶液一直在37 ℃下保温。通过分子模拟可知,与其它几种载体相比,Soluplus可以与Dio形成更强的氢键相互作用,应该能够有效抑制Dio结晶,通过抑晶实验来验证模拟结果。
由图2显示,Dio在无载体的溶出介质中快速沉淀,5 min后质量浓度低于检测限。Pluronic-F68和PEG4000在240 min内均不能抑制Dio在过饱和溶液中的结晶,PVP-K30的结晶抑制作用很弱。HPMC-E30具有一定的结晶抑制作用,虽然Dio在最初的15 min内迅速沉降在含有HPMC-E30的介质中,但仍可以保持一定的质量浓度。Soluplus对结晶的抑制作用最强,在含有Soluplus的过饱和溶液中,Dio的逐渐沉降和高质量浓度持续了4 h。结果表明,在过饱和溶液中,抑制结晶能力的大小顺序为Soluplus>HPMC-E30>PVP-K30>Pluronic- F68和PEG4000,无定形载体在抑制Dio结晶方面比半结晶载体更有效。
2.1.3 相图的绘制 药物与聚合物的混溶性是设计ASD最佳处方的关键参数,对于给定的药物-聚合物二元体系,其混溶性也是选择载药量的重要依据,关系到ASD的物理稳定性。混溶性决定了亚稳态ASD的载药上限,因为较高的载药量会导致自发相分离,致使药物结晶,这就否定了ASD的优势。混溶性曲线(即不同温度或组分下的混溶性)可以构建温度-组分相图,为ASD的配方设计和产品制造提供有价值的信息。事实上,根据F-H理论,热力学定义了旋节曲线,它是亚稳态的极限,在旋节曲线的任何一点上,都会发生称为旋节线分解的自发相分离。旋节曲线对应于混溶性曲线,可以根据F-H理论进行预测,得到相图[10]。对于药物-聚合物二元体系,混合的吉布斯自由能由公式(1)描述。
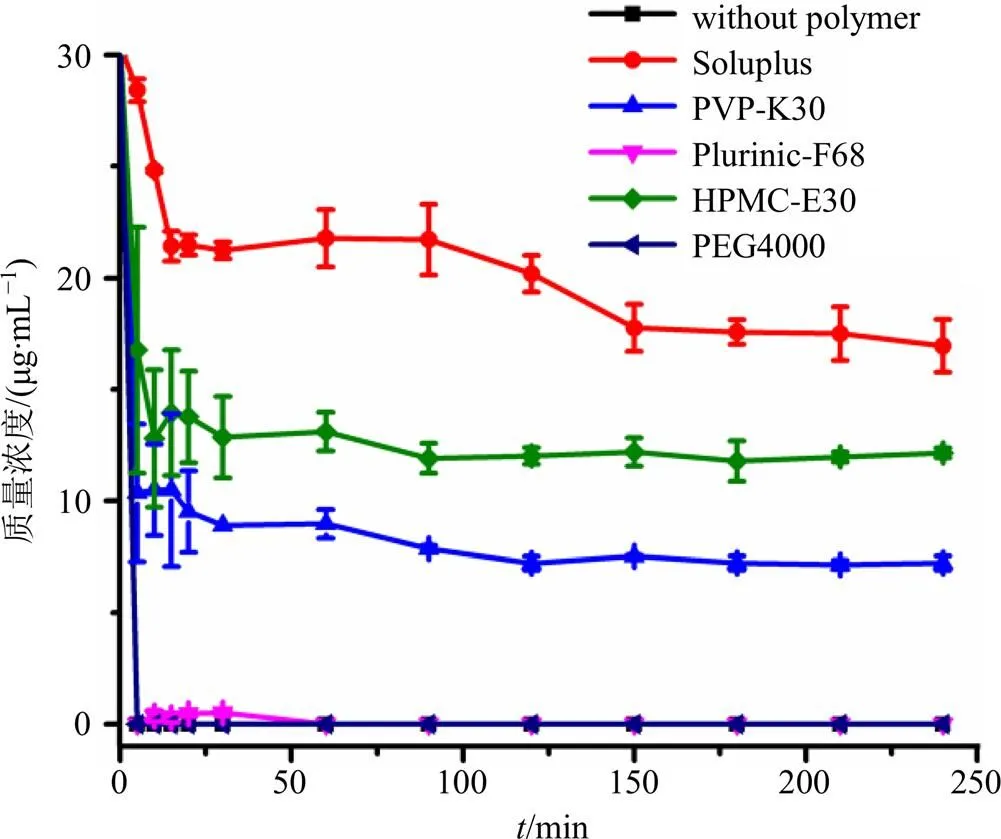
图2 在超饱和状态下不同聚合物对Dio的结晶抑制效应(, n = 3)
Δmix=R[ln+(1-)/ln(1-)+(1-)] (1)
R是气体常数,是绝对温度,是药物的体积分数,是被聚合物链占据的晶格位点数(在此定义为药物分子的体积),是药物-聚合物相互作用参数
聚合物链所占据的晶格位数和药物-聚合物相互作用参数可由公式(2)(3)计算。
=polymerdrug/drugpolymer(2)
=(drug-polymer)2/R(3)
为相对分子质量,是密度,是Hansen溶解度参数,是晶格点的体积(即药物的体积)
将自由能的二阶导数设为零,可以得到自旋点曲线,并表示为公式(4)。
S=2(drug-polymer)2/{R[1/+1/(1-)]} (4)
表1中示出了估计混溶性曲线(即旋节线曲线)所需的参数,根据公式(4)进行计算并得到混溶性曲线,结果见图3。
表1 Dio混溶性计算所需参数
Table 1 Data used for calculation of miscibility of Dio.
物质M/(g∙mol−1)ρ/(g∙cm−3)v/(cm3∙mol−1)δ/MPa1/2 Dio414.621.13366.817.21 Soluplus1180001.209833317.40
和的计算方法参考文献[11]
reference[11]for the calculation method ofand
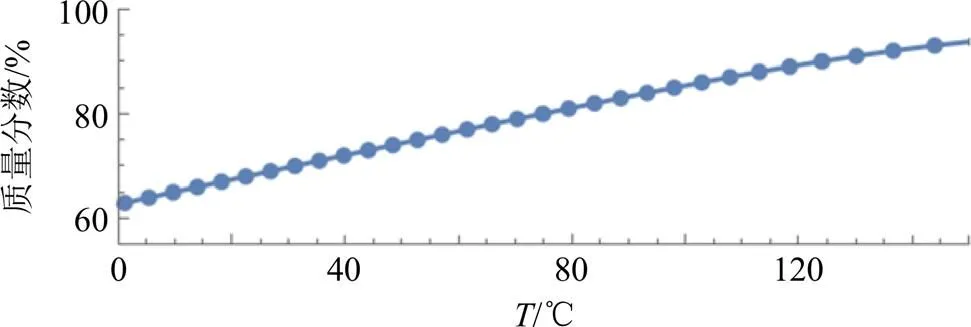
图3 预测Dio-Soluplus系统的温度-组分相图
由所构建的相图所示,在混溶性曲线之上的,将会发生自发相分离。从相图可以看出,混溶度随着温度的升高而增加,Dio和Soluplus在25 ℃下的混溶性68.57%,在室温下ASD的最大载药量不能超过68.57%。
2.2 Dio-ASD制备工艺优化
2.2.1 色谱条件 色谱柱为Diamonsil Plus C18柱(150 mm×4.6 mm,5 μm);流动相为乙腈-水(84∶16);检测波长203 nm;柱温30 ℃;体积流量1 mL/min。
2.2.2 物理混合物(physical mixture,PM)的制备 按一定比例精确称量已经研磨过80目筛的原料药和Soluplus,装到真空袋中,混匀,即得。
2.2.3 溶出度测定 取Dio、Dio-ASDs及PM,按照《中国药典》2020年版溶出度与释放度测定方法中“浆法”进行溶出实验,以900 mL 0.1% SDS溶液为介质,在37 ℃、100 r/min下试验,分别于5、15、30、45、60、90、120 min取样5 mL,经0.45 μm微孔滤膜滤过后测定,同时补充同温等量介质计算得到溶出度。
2.2.4 共沉淀法制备Dio-ASD 以Soluplus为载体,按原料药与载体的比例(药载比)1∶2、1∶4、1∶6、1∶8、1∶10、1∶12精确称量,加无水乙醇超声至完全溶解,旋转蒸发挥干溶剂后冷冻30 min,放50 ℃烘箱干燥过夜,取出粉碎,过80目筛,测定溶出度,结果见图4。可见在药载比为1∶10时,溶出度最高,此时的载药量为9.10%,没有超过按混溶性曲线计算的最大载药量68.57%,推测此时药物和载体应具有较好的混溶性,因此,选择此比例为最佳工艺。
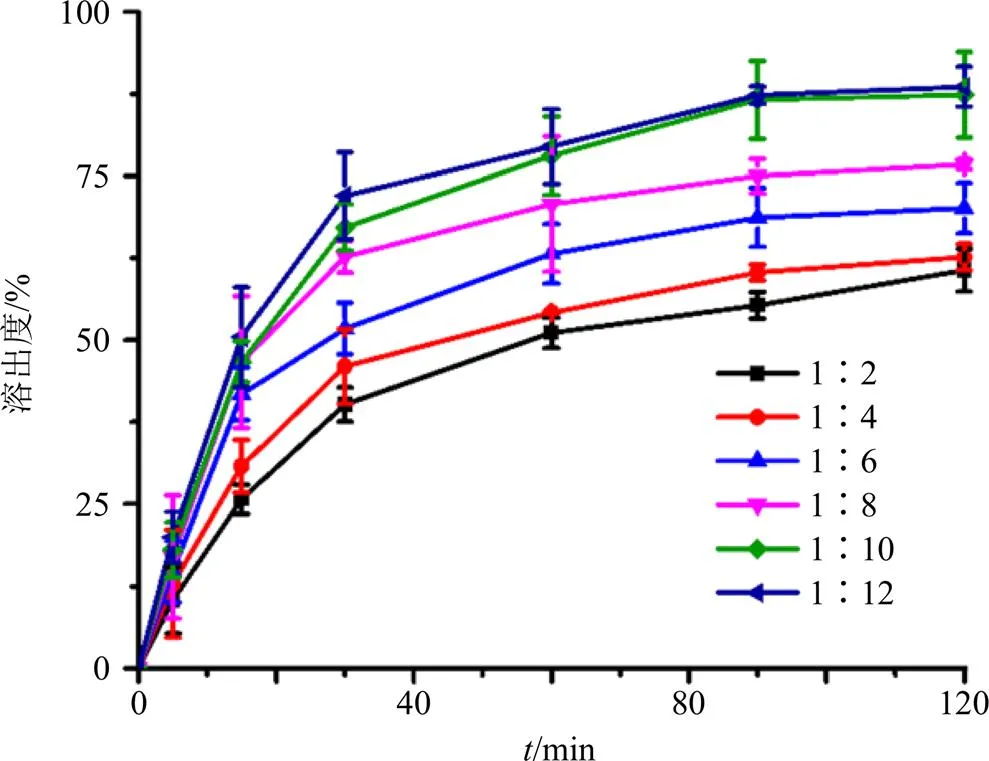
图4 Dio-ASDs中不同Dio/Soluplus比例的溶出度(, n = 3)
2.2.5 工艺验证 按优选的最佳比例制备Dio- ASDs和PM各3批,测定溶出度,结果见图5。在0.1% SDS溶液中,Dio-ASDs的溶出度明显高于原料药Dio和PM,原因可能是药物在溶出介质中润湿性的改善[12]。
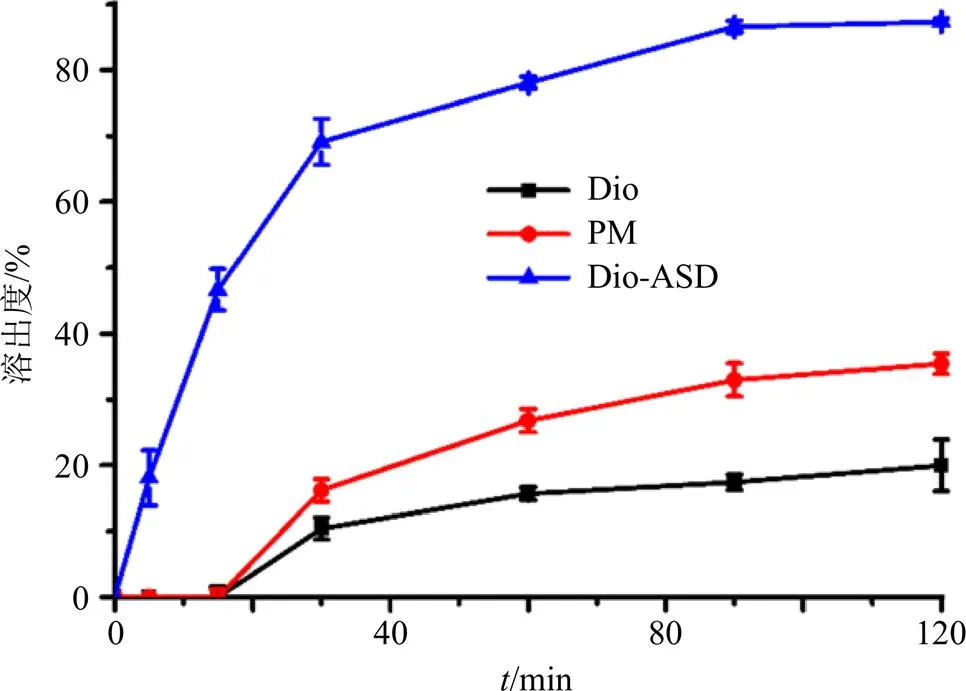
图5 药物与载体的质量比为1∶10时Dio-ASD的溶出度(, n = 3)
2.3 Dio-ASD物相表征
2.3.1 DSC分析 取待测样品约5 mg,精密称定,装入铝坩埚中,以空坩埚为参比,进行DSC分析。工作条件:扫描速度10 ℃/min,N2吹扫气体积流量40 mL/min,N2保护气体积流量60 mL/min,扫描范围25~300 ℃。DSC图谱如图6所示。图6显示了Dio、Soluplus、PM和Dio-ASD的热行为。在213.67 ℃时,Dio出现了1个明显的吸热峰,表明Dio原料药为晶体结构。Dio-ASD中Dio的吸热峰消失,证实该药物与载体混溶,药物成无定形态。本实验PM未发现明显吸热峰,可能是由于在DSC程序升温时使先熔融的载体成为Dio良好的溶剂,使其在到达熔点之前就已逐渐溶解在熔融的载体中,或者只是药物的结晶性受到Soluplus的抑制,由于DSC灵敏度有限而并未检测到明显吸热峰,因此,并不能确定PM中药物以无定形态存在,需要通过PXRD和SEM进一步确证。
2.3.2 X-射线粉末衍射(X-ray powder diffraction,XRPD)分析 分析条件为Cu靶,波长为0.154 0 nm,管流强度40 mA,管电压40 kV,扫描角度10°~60°,步长0.02°,扫描频率8°/min。对样品进行XRPD分析。图7显示了Dio、Soluplus、PM和Dio-ASD的XRPD图像。Dio在5°~20°有特征的晶体衍射峰,分别位于7.14°、14.22°、14.90°、16.08°、16.98°、17.78°、18.64°,在PM衍射图中可以清楚地观察到Dio的所有主要特征晶体峰,Dio-ASD中的Dio晶体特征峰消失,表明载体与Dio之间的相互作用可以有效地将药物的晶体状态转变为无定形态,此结果与DSC结果一致。
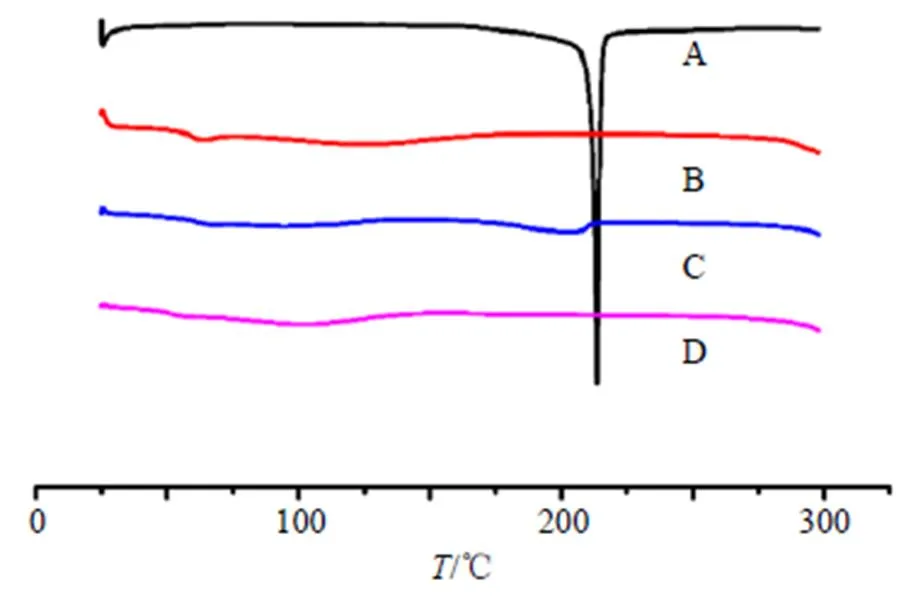
图6 Dio (A)、Soluplus (B)、PM (C)和Dio-ASD (D)的DSC图谱
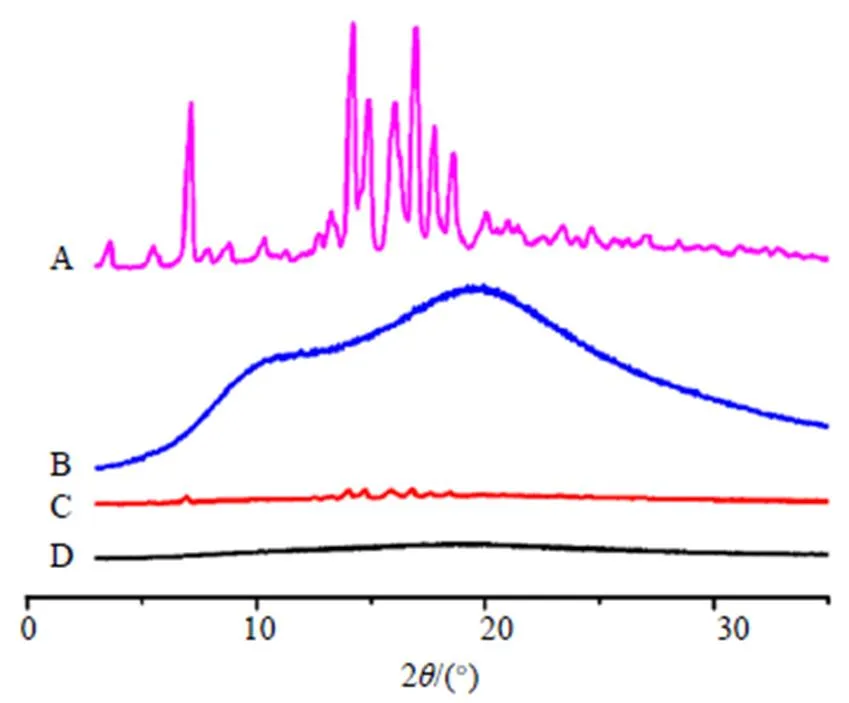
图7 Dio (A)、Soluplus (B)、PM (C)和Dio-ASD (D)的XRPD图谱
2.3.3 SEM观察 将样品涂于干净的铜片上,喷金后观测各样品的表面形态。结果如图8所示。Dio呈棒状或粒状结晶结构,Soluplus以无定形态存在,能观察到PM中Dio晶体出现在载体中,而Dio-ASD中没有Dio晶体结构,这一结果表明Dio以无定形形式存在于ASD体系中。

图8 Dio (A)、Soluplus (B)、PM (C)和Dio-ASD (D)的SEM图谱
2.3.4 FT-IR分析 采用KBr压片法将样品压片,在4000~400 cm−1扫描范围分别进行FT-IR分析。结果如图9所示。Dio有如下特征峰(cm−1):3 452.2(-OH)、1 241.5、1 053.1(3β-OH,∆5)、980.4、峰强度918.7<897.4、866.3(25R螺甾烷);Soluplus存在-OH、O=C-S-和O=C-C=C-羰基的伸缩振动特征峰,分别位于3 465.3、1 739.2、1 637.7 cm−1;PM与Dio红外光谱图基本相似,Dio的特征峰基本都存在,说明药物和载体之间只是简单混合,并没有发生相互作用;相反,在Dio-ASD中来自Dio的羟基峰发生了位移,提示药物和载体之间可能存在氢键。Dio特征峰3 452.2 cm−1(-OH)的小尖峰消失,与Soluplus的羟基峰合并为1个宽、钝的吸收峰,位于3 459.8 cm−1。-OH的消失表明Dio的羟基与Soluplus的羰基(氢键受体)之间存在分子间氢键。值得注意的是,药物和载体之间的相互作用对ASD是一个额外的好处,因为它们不仅可以抑制药物的结晶,而且可以增强药物在亲水性辅料中的固溶度[13]。
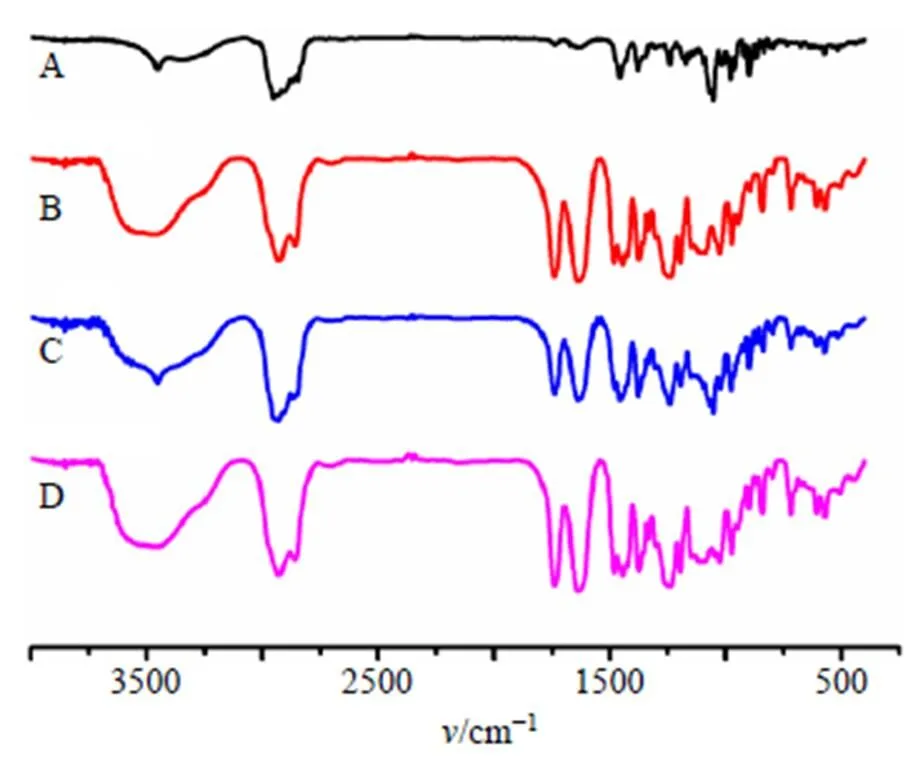
图9 Dio (A)、Soluplus (B)、PM (C)和Dio-ASD (D)的FT-IR图谱
2.4 Dio-ASD药动学研究
2.4.1 给药方案 将SD大鼠随机分组,每组6只。分别口服Dio、PM和Dio-ASD,给药剂量为100 mg/kg。给药后于0.083、0.25、0.5、0.75、1、2、3、4、6、8、12、24、36、48、72 h大鼠眼眶取血,置于肝素处理的EP管中,离心(转速为10 000 r/min)10 min,即得血浆,迅速将血浆样品保存于−80 ℃冰箱。利用UPLC-MS/MS法测定血浆中Dio的血药浓度。
2.4.2 分析方法及样品处理方法
(1)色谱条件:色谱柱:Waters Acquity UPLC®BEH C18柱(50 mm×2.1 mm,1.7 μm);流动相为乙腈-0.03%甲酸水溶液(80∶20);柱温35 ℃;样品温度4 ℃;进样量2 μL;体积流量0.3 mL/min。
(2)质谱条件:离子源参数如下:ESI(+);Dio去簇电压71.96 V,碰撞电压23.03 eV;丹参酮IIA(内标)去簇电压120 V,碰撞电压25.92 eV;用于定量分析的离子对如下:/415.2→271.2(Dio);/295.2→277.1(丹参酮IIA)。
(3)方法学考察:参照《中国药典》2020年版附录生物样品定量分析方法验证指导原则,对专属性、线性范围、精密度与准确度、提取回收率与基质效应和稳定性进行方法学考察,所有项目考察结果均符合规定。
(4)血浆处理:精密量取50 μL大鼠血浆,加入50 μL内标(41.85 ng/mL),涡旋30 s,加入250 μL甲醇-乙腈(1∶1),涡旋2 min,13 000 r/min高速离心15 min,取上清液,UPLC-MS/MS分析,药时曲线结果见图10,药动学参数见表2。
2.4.3 数据处理与药动学参数计算 实验前取出血浆,在室温下融化后按“2.4.2”项下分析方法进行检测,收集实验数据。采用SPSS 19.0统计软件对试验数据进行处理,计量资料以表示,各组间比较釆用单因素方差分析(One-way ANOVE)。使用DAS 3.0药动学处理软件,选择非房室模型方法进行药动学参数计算。由图10和表2可知,Dio-ASD的AUC0~t和max分别是Dio的5.84、3.17倍。
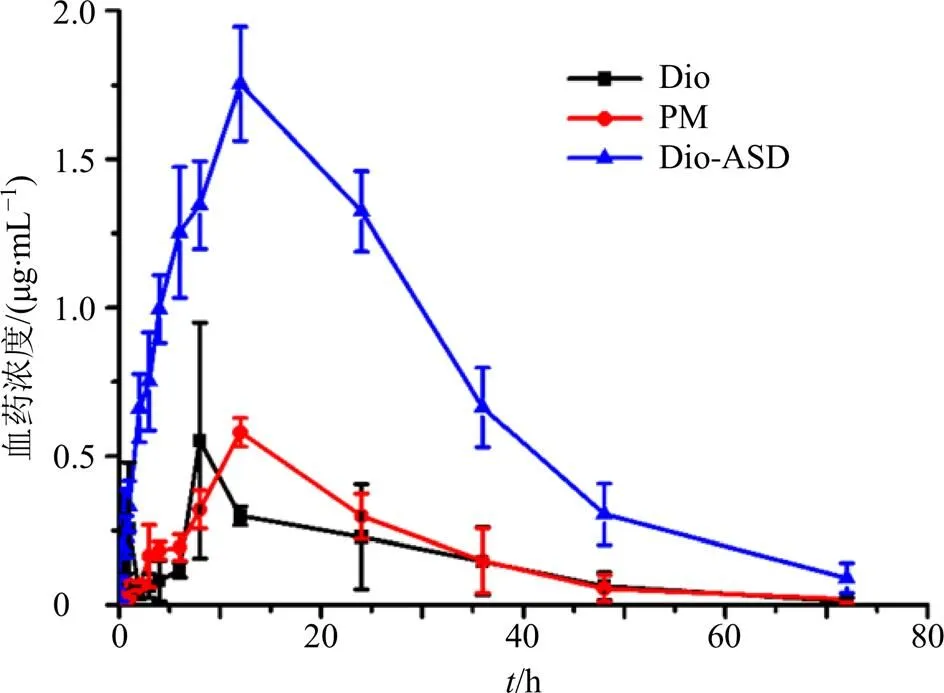
图10 口服Dio和Dio-ASD后Dio的血药浓度-时间分布(, n = 6)
表2 口服Dio and Dio-ASD后药动学参数(, n = 6)
Table 2 Pharmacokinetic parameters following oral administration of Dio and Dio-ASD (, n = 6)
参数单位DioPMDio-ASD AUC0~tμg∙h∙L−19 302.24±4013.3812 101.45±3 357.2654 386.54±8 864.42** t1/2zh12.33±3.029.11±2.2910.37±1.68 tmaxh8.37±2.5412.16±4.9210.98±1.99 Cmaxμg∙L−1552.63±100.13567.88±88.341 742.65±491.46**
与Dio比较:*<0.05**<0.01
*< 0.05**< 0.01Dio
应用下列公式计算Dio-ASD的相对生物利用度()。
=AUC0~∞ Dio-ASD/AUC0~∞ Dio
计算得Dio-ASD的相对生物利用度分别为589.47%(<0.05),即采用共沉淀法将Dio制备成ASD后,Dio呈现无定形状态,溶出度增加,其生物利用度有了很大的提高。
2.5 Dio-ASD稳定性研究
样品平铺于培养皿中,厚度约2~3 cm,保存在40 ℃,75% RH下,于0、3、6个月时分别取样,进行XRPD测定,来检测形态随时间的变化,并采用HPLC对不同贮藏时间的体外溶出度进行测定。结果见图11、12。如XRPD衍射峰图(图11)所示,存储后的Dio-ASD与初始状态相似,没有显示Dio的任何衍射峰。在40 ℃,75%的湿度下保存6个月后溶出度(图12)与新制备的ASD相比无显著性差异。进一步证实了该无定形药物不结晶并有良好的稳定性。
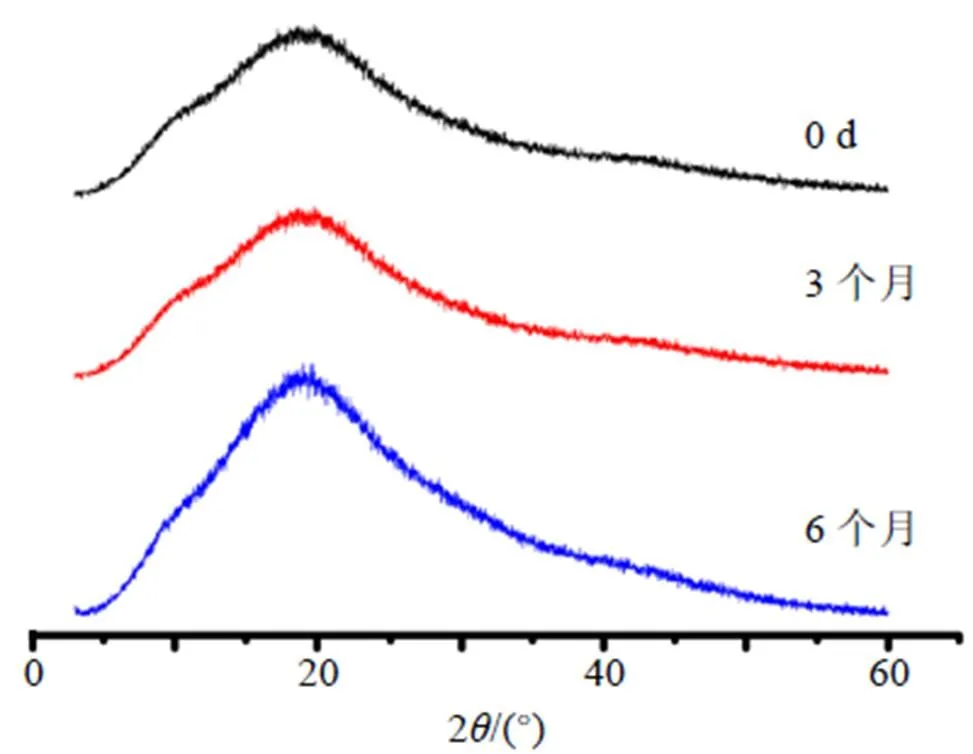
图11 Dio-ASD放置0、3、6个月后的XRPD图谱
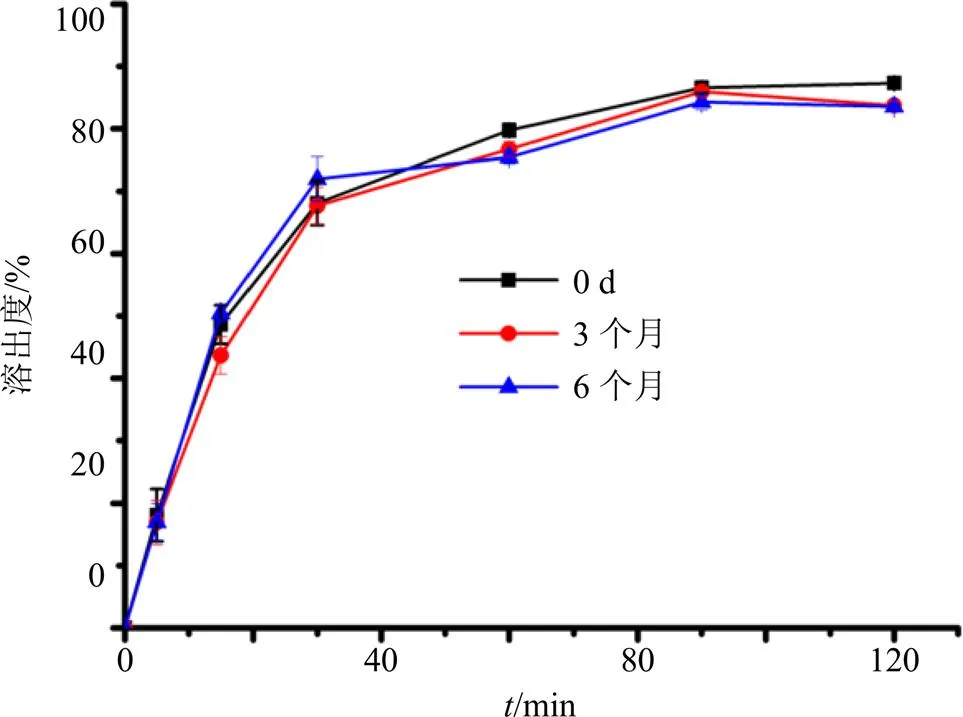
图12 Dio-ASD放置0、3、6个月后Dio的溶出度测定结果(, n = 3)
3 讨论
分子模拟是将量子力学理论与计算方法相结合研究分子结构的一项强有力的技术。它可以在分子力学和量子化学理论的基础上模拟分子在原子水平上的行为。将势函数和力参数描述为力场,计算相互作用的分子间的力和整个系统的总能量。分子模拟可以提供可见的三维结构和轨迹剖面。这些参数(如自由能和动态参数)可以从轨迹中计算出来,从而揭示分子结构[14]。由于分子建模的独特优势,在药物研究中的应用实例越来越多。文献报道采用模拟退火法研究了一系列布洛芬ASD与聚乙二醇、泊洛沙姆、聚维酮、乳糖和甘露醇相互作用的分子结构,并用分子动力学模拟方法研究了布洛芬十二烷基硫酸钠与聚乙二醇、聚维酮和泊洛沙姆的溶解过程[15]。因此,分子模拟是在分子水平上解释实验现象的重要工具。本实验中,由于氢键和疏水作用,Dio和Soluplus之间形成了很强的结合能。Dio和Soluplus之间分子相互作用可有效抑制药物结晶,进而提高其溶解性和稳定性。
ASD已被证明是提高低水溶性药物溶解度、溶出度和生物利用度最有前景的策略之一。然而,ASD的配方设计仍然是药学工作者面临的一大挑战。研究表明,晶体药物在无定形聚合物中的溶解度和药物与聚合物的相溶性是设计最佳处方的2个关键参数。对于给定的药物-聚合物二元体系,其溶解度和混溶性是选择载药量的重要依据,这关系到ASD的物理稳定性[16]。混溶性决定了亚稳态ASD的载药上限,因为较高的载药量会导致自发相分离,药物结晶,这就否定了ASD的优势。此外,玻璃化转变温度(g)是ASD的另一个关键参数[17]。溶解度曲线、混溶性曲线和g曲线(即不同温度或组分下的溶解度、混溶性和g)可以构建温度-组分相图,为ASD的配方设计和产品制造提供有价值的信息。然而,由于聚合物的高黏度,特别是在低于其g的温度下,达到溶解度平衡所需的时间相当长,这使得在各种温度下确定药物-聚合物的溶解度和可混溶性成为一个挑战。因此,在这种情况下,理论预测是有利的[18]。因此,本实验通过绘制相图理论预测药物和聚合物之间的混溶性,推测出制备的ASD载药上限,为后续实验提供了理论依据,具有一定的理论创新性。
在口服给药领域,通过过饱和增加腔内浓度有望增强肠道吸收[19-20]。加入聚合物载体可以稳定和延长过饱和度,其对ASD中无定形态药物结晶抑制机制包括与药物在局部产生的相互作用、无定形态药物的分子流动性降低和药物结晶成核活化能及g的提高。在此,本课题组进行了高通量沉淀筛选来选择载体,并在筛选/优化过程中评估过饱和潜力[21-22]。在本实验中,采用超饱和溶液法研究了5种不同水溶性载体对Dio晶体的抑制效果,无定形载体在抑制Dio结晶方面比半结晶载体更有效。Soluplus对Dio的抑晶效果最佳,原因可能是其水溶性、黏度及三维结构体系降低了药物的自由能,减缓过饱和溶液中药物的析晶沉淀,起到维持过饱和度的作用。值得注意的是,Soluplus在纯水中的CMC仅为7.6 mg/L,在不同PBS(pH 6.8、6.5、7.0)溶液中,CMC值分别为0.61、1.24和0.91 μg/mL,在0.1 mol/L HCl(pH 1.0),Soluplus的CMC值为11.7 μg/mL[23],这些研究结果证实Soluplus在溶液中极易形成胶束,这种增溶作用不仅可以间接地促进过饱和体系的形成,而且作为一个有利于快速补充及时吸收的游离药物的药物库。此外,前期预实验过程中,我们在药物与载体的比例为1∶3时,观察了不同载体对Dio的抑晶作用。结果表明,除Soluplus具有微弱的晶体抑制作用外(Dio的质量浓度可在0.003 mg/mL下维持2.5 h),所有其他载体均无晶体抑制作用。这说明载体浓度的增加对抑制Dio结晶有有利的影响。而不同药载比下(1∶1、1∶3、1∶6、1∶9、1∶12)Soluplus对Dio的抑晶作用结果显示药载比在1∶6和1∶9时呈现最佳效果(二者相差无几),为节约材料,选择1∶6。
Soluplus是巴斯夫公司用于热熔挤出技术而生产的,本实验所用的共沉淀法为Soluplus的应用扩展。由DSC、XRPD、SEM和FT-IR等结果表明Dio-ASD中的Dio以无定形形式存在,药物和载体之间存在相互作用,而药物润湿性的改善、从结晶状态到无定形的转变[24]和药物载体分子间的氢键效应都有利于增强Dio的溶出,进而提高其生物利用度。
利益冲突 所有作者均声明不存在利益冲突
[1] 张释晴, 宋雨轩, 张文雪, 等. 抗肿瘤天然产物薯蓣皂苷元的研究进展 [J]. 中国中药杂志, 2021, 46(17): 4360-4366.
[2] 王洋, 赵立春, 庞宇舟, 等. 不同剂量薯蓣皂苷元的大鼠体内药动学研究 [J]. 中国医院药学杂志, 2020, 40(3): 269-273.
[3] Okawara M, Tokudome Y, Todo H,. Effect of β-cyclodextrin derivatives on the diosgenin absorption in Caco-2 cell monolayer and rats [J]., 2014, 37(1): 54-59.
[4] Okawara M, Tokudome Y, Todo H,. Enhancement of diosgenin distribution in the skin by cyclodextrin complexation following oral administration [J]., 2013, 36(1): 36-40.
[5] Liu C Z, Chang J H, Zhang L,. Preparation and evaluation of diosgenin nanocrystals to improve oral bioavailability [J]., 2017, 18(6): 2067-2076.
[6] Okawara M, Hashimoto F, Todo H,. Effect of liquid crystals with cyclodextrin on the bioavailability of a poorly water-soluble compound, diosgenin, after its oral administration to rats [J]., 2014, 472(1/2): 257-261.
[7] Prasad E, Robertson J, Halbert G W. Mefenamic acid solid dispersions: Impact of formulation composition on processing parameters, product properties and performance [J]., 2022, 616: 121505.
[8] Chavan R B, Rathi S, Sainaga Jyothi V G S,. Cellulose based polymers in development of amorphous solid dispersions [J]., 2019, 14(3): 248-264.
[9] Chen J, Ormes J D, Higgins J D,. Impact of surfactants on the crystallization of aqueous suspensions of celecoxib amorphous solid dispersion spray dried particles [J]., 2015, 12(2): 533-541.
[10] Tian B, Wang X Y, Zhang Y Y,. Theoretical prediction of a phase diagram for solid dispersions [J]., 2015, 32(3): 840-851.
[11] Forster A, Hempenstall J, Tucker I,. Selection of excipients for melt extrusion with two poorly water-soluble drugs by solubility parameter calculation and thermal analysis [J]., 2001, 226(1/2): 147-161.
[12] Vasconcelos T, Marques S, das Neves J,. Amorphous solid dispersions: Rational selection of a manufacturing process [J]., 2016, 100: 85-101.
[13] Riekes M K, Kuminek G, Rauber G S,. HPMC as a potential enhancer of nimodipine biopharmaceutical properties via ball-milled solid dispersions [J]., 2014, 99: 474-482.
[14] de Vivo M, Masetti M, Bottegoni G,. Role of molecular dynamics and related methods in drug discovery [J]., 2016, 59(9): 4035-4061.
[15] Chan T, Ouyang D F. Investigating the molecular dissolution process of binary solid dispersions by molecular dynamics simulations [J]., 2018, 13(3): 248-254.
[16] Zhao Y Y, Inbar P, Chokshi H P,. Prediction of the thermal phase diagram of amorphous solid dispersions by flory-Huggins theory [J]., 2011, 100(8): 3196-3207.
[17] Kaushal A M, Gupta P, Bansal A K. Amorphous drug delivery systems: Molecular aspects, design, and performance [J]., 2004, 21(3): 133-193.
[18] Bellantone R A, Patel P, Sandhu H,. A method to predict the equilibrium solubility of drugs in solid polymers near room temperature using thermal analysis [J]., 2012, 101(12): 4549-4558.
[19] Pas T, Bergonzi A, Lescrinier E,. Drug-carrier binding and enzymatic carrier digestion in amorphous solid dispersions containing proteins as carrier [J]., 2019, 563: 358-372.
[20] Sun D D, Lee P I. Crosslinked hydrogels-a promising class of insoluble solid molecular dispersion carriers for enhancing the delivery of poorly soluble drugs [J]., 2014, 4(1): 26-36.
[21] Chavan R B, Rathi S, Jyothi V,. Cellulose based polymers in development of amorphous solid dispersions [J]., 2019, 14(3): 248-264.
[22] Bevernage J, Forier T, Brouwers J,. Excipient-mediated supersaturation stabilization in human intestinal fluids [J]., 2011, 8(2): 564-570.
[23] Shi N Q, Lai H W, Zhang Y,. On the inherent properties of Soluplus and its application in ibuprofen solid dispersions generated by microwave-quench cooling technology [J]., 2018, 23(6): 573-586.
[24] Bikiaris D N. Solid dispersions, part I: recent evolutions and future opportunities in manufacturing methods for dissolution rate enhancement of poorly water-soluble drugs [J]., 2011, 11(8): 1501-1519.
Preparation and evaluation of diosgenin amorphous solid dispersionand
LIU Pei, CHANG Jin-hua,KANG Kai, XUE He-fei, WANG Yu-xin, XU Lin, LIU Cui-zhe, ZHOU Jian-yu
Hebei Province Key Laboratory of Nerve Injury and Repair, Hebei Province Key Laboratory of Research and Development for Chinese Medicine, Chengde Medical College, Chengde 067000, China
To prepare diosgenin amorphous solid dispersion (Dio-ASD) and improve the dissolution and bioavailability of Dio.The interaction between Dio and carrier was analyzed by molecular simulation technology and verified by crystal suppression experiment. The phase diagram of the miscibility curve between Dio and carrier was constructed, and the miscibility of Dio and carrier was predicted theoretically. Dio-ASD was prepared by coprecipitation with Soluplus as carrier and was evaluatedby dissolution assay, differential scanning calorimetry (DSC), X-ray powder diffraction (XRPD), scanning electron microscopy (SEM), and fourier transform infrared spectroscopy (FT-IR). The plasma concentration of Dio in rats was determined by UPLC-MS/MS method, and the pharmacokinetic parameters were calculated to evaluate Dio-ASD.Molecular simulation results showed that hydrophobic and hydrogen interactions between Soluplus and Dio were formed, and the binding energy was stronger compared with other carriers. Soluplus had the strongest crystal suppression effect on Dio. The phase diagram of the miscibility curve was constructed and the miscibility between Dio and Soluplus at 25 ℃ was 68.57%. Compared with the bulk drug, the dissolution rate of Dio-ASD was significantly improved. The phase characterization results showed that Dio existed in Dio-ASD in an amorphous state, and there was an interaction between Dio and Soluplus. The pharmacokinetics results showed that the bioavailability of Dio-ASD was nearly five times higher than that of Dio after intragastric administration to rats.The prepared Dio-ASD can significantly improve drug dissolution and bioavailability in rats.
diosgenin; amorphous solid dispersion; molecular simulation; miscibility phase diagram; pharmacokinetics
R283.6
A
0253 - 2670(2022)14 - 4323 - 10
10.7501/j.issn.0253-2670.2022.14.012
2022-02-09
河北省高等学校科学技术研究项目(QN2020241);河北省高等学校科学技术研究项目(QN2019167);河北省高校重点学科建设项目(冀教高[2013]4号);河北省科技厅“技术创新引导专项-科技工作会商”项目;河北省神经损伤与修复重点实验室开放课题(NJKF202102)
刘 沛(1985—),女,博士,副教授,研究方向为药物新剂型及药动学研究。Tel: (0314)2290629 E-mail: liupeipp123@163.com
刘翠哲(1964—),女,博士,博士生导师,研究员,研究方向为中药制剂现代化及中药药动学研究。Tel: (0314)2517035 E-mail: liucuizhexy@163.com
周剑宇(1983—),男,博士,硕士生导师,副教授,研究方向为中药药效与物质基础研究。Tel: (0314)2290076 E-mail: zhoujybucm@163.com
[责任编辑 郑礼胜]
