Discovery Studio软件在兽医药物化学中的应用实践
2022-05-16王战辉张素霞吴聪明沈建忠
王战辉,张素霞,吴聪明,沈建忠
(中国农业大学动物医学院,北京 100193)
药物化学指的是应用化学和生物学的概念及方法发现、确证和开发新药,从分子水平上研究药物的作用机制及其在体内的作用方式。药物化学的内容涉及发现、修饰和优化先导化合物,从分子水平上揭示药物及具有生理活性物质的作用机理,研究药物及生理活性物质在体内的代谢过程。近代药物化学的发展非常迅速。药物化学家们一直在研究如何能够通过合理药物设计而发现新药,在计算机科学、结构生物学等相关学科高速发展的今天,特别是计算机辅助药物分子设计(computer aided drug design,CADD)的完善,利用计算机模拟、高效合成技术可以加快新药发现的速度,降低新药开发的成本[1]。CADD已经成为当前药物合理设计中不可或缺的环节。
据统计,药物作用的靶点中最多的是受体,其次是酶,另外还有离子通道和核酸,但占比较少。尽管药物作用靶点可能不同,但是药物的作用方式都是以非键相互作用为主,比如疏水作用力、氢键、范德华力等[2]。相对于比较直观的原子与原子之间的共价键、离子键等,非键相互作用要更加抽象。目前,主流的蛋白质三维结构解析方法有X射线晶体衍射法、核磁共振法和冷冻电镜法3种。X射线晶体衍射法的前提是获得蛋白质晶体,然后通过X射线衍射解析蛋白的三级结构,这种方法分辨率较高,一般能做到0.3 nm以内。核磁共振法要求蛋白分子量要小且稳定,适用范围要小于X射线衍射法。近些年发展比较迅速的冷冻电镜法有其得天独厚的优势,适合用于解析病毒、细胞等生物结构中较大的蛋白质,但也有分辨率不高,不适合解析较小蛋白质的缺点。目前,通过冷冻电镜解析的最小蛋白质为52 ku,分辨率仅有0.32 nm[3]。总体而言,蛋白质的三维结构解析的速度较慢,成本较高,很多时候不能满足科研的需要。因此,基于蛋白的一级结构建立其三维结构模型就成为一种非常重要的计算机模拟技术。
随着蛋白质建模技术的发展,建模方法逐渐归为两种,分别是从头设计和同源建模。从头设计依赖强大的计算资源和复杂的算法,应用没有同源建模广泛。同源建模是在有相似蛋白质模板的情况下构建目标序列的三维结构模型的方法,而且是目前使用最广泛、预测最可靠的建模方法[4]。通过建立蛋白质的三维结构模型,对于深入研究药物与靶点之间的非键相互作用具有重要的意义,为药物的合理设计提供理论基础。获得蛋白质的模型后,就可以对其进行分子对接,分析受体-配体之间的相互作用。分子对接的产生可以追溯到19世纪Fisher提出的受体学说,最经典的模型就是锁钥模型[5]。随着受体学说的发展,人们对药物分子与靶点的相互作用有了更深入的认识。目前,基于药物分子与靶点之间的空间匹配和能量匹配的分子对接方法主要分为刚性对接、半柔性对接和柔性对接3类[6]。刚性对接计算量相对较小,参与对接的药物分子和受体的构象均不发生变化,计算速度较快,但是计算精度较差,一般用于大分子对接或大量对接初步筛选等。柔性对接需要的计算量较大,该方法需要同时把药物分子和受体构象的变化考虑进去,因此,适合精确的分子对接。半柔性对接需要的计算量适中,一般固定受体的构象,然后考虑药物分子的结构变化,该方法的计算结果相对准确,需要的计算资源又不是特别巨大,是应用最广泛的对接方法之一。常用的分子对接软件有Discovery Studio、Sybyl、MOE、Rosetta、Schrodinger、AutoDock等[7]。
在本文中,选择Discovery Studio软件中的同源建模模块构建磺胺类药物受体蛋白DHPS的三维结构模型,然后使用半柔性对接方法CDOCKER进行受体-配体的分子对接,通过二维平面图以及三维结构图展示药物分子与受体之间的相互作用方式,借助直观的模拟展示,以提升学习药物化学的效果。
1 研究方法和原理
1.1 Discovery Studio简介
Discovery Studio(DS)是基于Windows/Linux系统的分子建模和模拟软件,在药物发现、药物结构功能研究等领域应用广泛。DS软件界面友好,功能完善,易于学习,是计算机辅助药物设计最常用的模拟软件之一[8]。DS涵盖疾病的发病机理研究、新药发现和设计、计算药物化学、生物信息学、结构生物学、免疫学、病毒学、肿瘤药物研究等研究领域。功能模块包括基于结构的药物发现和设计模块、基于片段的药物设计模块、基于药效团的药物发现和设计模块、基于小分子的药物发现和设计模块、蛋白质建模及模拟模块、分子力学、分子动力学和量子力学模拟模块等。利用DS软件学习药物化学,可直观地观察配体与受体间的结合位点,并将两者间的非键相互作用以图形的方式展示,可对分子间相互作用有感性认识,加深对受体-药物相互作用的理解。DS有单机版本和免费的Discovery Studio Visualizer模块,可随时利用个人电脑自主练习,加深学习印象,激发对抽象概念的兴趣。
1.2 蛋白质同源建模
磺胺类药物受体蛋白二氢蝶酸合成酶(DHPS)是研究磺胺类药物关于细菌耐药产生的影响[9]和基于此受体的新型抗菌药物的关键蛋白。科研工作者们测定了多种来源的DHPS基因序列,并通过蛋白结晶和分子模拟技术等获得了受体的三维结构,目前,Protein Data Bank中已经收录了包括6种细菌来源、1种真菌来源的DHPS晶体结构。
DHPS的氨基酸序列可以通过文献或者NCBI数据库获得。以蛋白3tyz为例,通过NCBI下载FASTA格式的氨基酸序列文件并用DS软件打开,展开Macromolecules下的Create Homology Models模块,点击BLAST Search进行模板搜索,选择氨基酸序列一致性75%的lajoA作为模板蛋白,通常认为一致性大于60%则模板蛋白质量较好。点击Load Structure and Aligment进行模板蛋白的下载和序列比对(如图1所示)。点击Create Homology Models模块下的Build Homology Models,以lajoA作为模板蛋白构建蛋白三维模型(如图2所示)并采用拉氏构象图和Profile-3D对其进行评估。拉氏构象图通过选取主菜单Chart下的Ramachandran Plot显示,Profile-3D通过点击Create Homology Models模块下的Verify Protein(Profile-3D)进行计算。

图1 模板1ajoA与3tyz的序列比对结果
1.3 受体-配体分子对接
多种来源的DHPS已经被解析出了晶体结构[10],研究发现,其结合位点相对固定,极大方便了对接活性位点的选择。如果结合位点不能确定,则需要进行氨基酸突变来试验确认。
1.3.1 配体分子的构建 以磺胺甲噁唑SMZ作为分子对接的配体分子。使用ChemDraw软件绘制SMZ结构并导入到DS软件中,展开Small Molecules下的Minimize Ligands模块,点击Full Minimization进行小分子结构批量优化。配体分子的获得途径有很多种,对于常见的配体分子可以直接从公开的Pubchem数据库中获得其结构,对于不常见的配体分子则可以通过ChemOffice或DS软件直接绘制,再通过DS进行简单优化即可,当然也可以通过高斯软件对其进行量子力学优化[11],获得其最低能量构象。

图2 蛋白3tyz模型(a)、拉氏构象图(b)及其Profile-3D图(c)
1.3.2 定义结合位点以及分子对接 要进行分子对接,首先要定义结合位点。展开DS软件中Receptor-Ligand Interactions下的Define and Edit Binding Site模块,点击From Receptor Cavities,可获得多个活性位点,由于磺胺类药物受体蛋白DHPS的活性口袋相对固定,选择与以往研究和实验相符的口袋作为分子对接结合位点。确定结合位点之后进行分子对接,展开Receptor-Ligand Interactions下的Dock Ligands模块,点击Dock Ligands(CDOCKER),选好受体、配体和活性位点点击确定进行对接计算。对接完成后,展开Receptor-Ligand Interactions下的View Interactions,点击Ligand Interactions,即可展示受体-配体相互作用关系。点击Show 2D Diagram,就可以看到平面的受体-配体互作图(如图4所示),点击Hydrophobic,可以展示受体疏水作用表面(如图5a所示);点击H-bond,可以展示受体氢键表面(如图5b所示)。
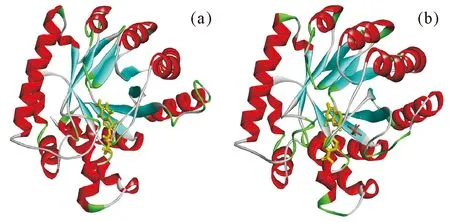
图3 3tyz蛋白模型与配体分子的对接构象图(a)和3tyz蛋白晶体复合物图(b)

图4 受体-配体相互作用平面图
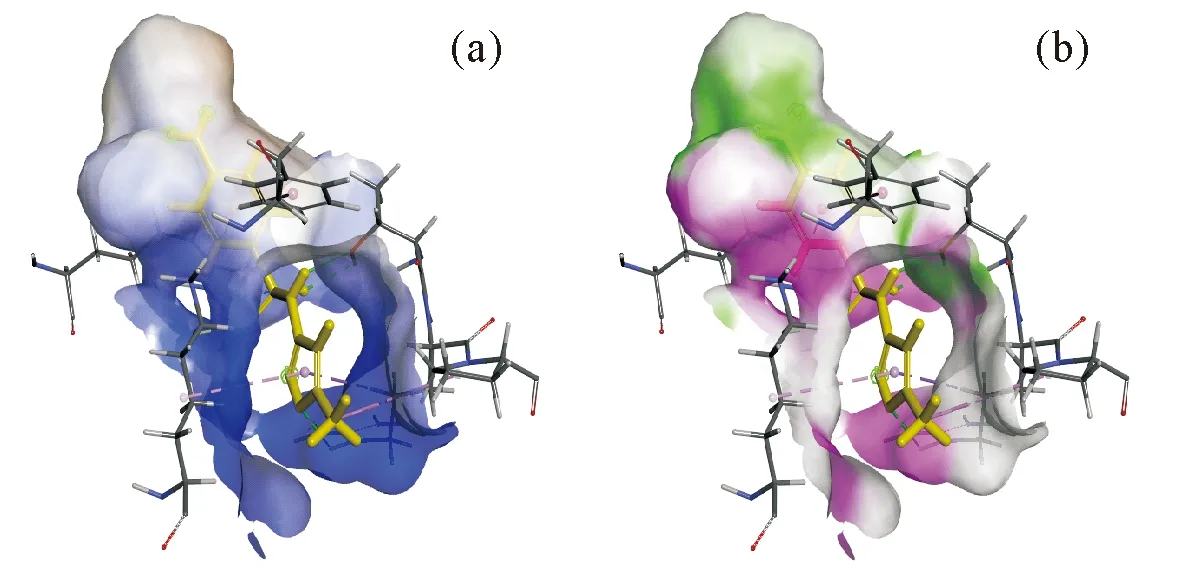
图5 受体疏水作用力表面图(a)和氢键作用力表面图(b)
2 结果
2.1 DHPS序列比对、同源建模与评估
3tyz氨基酸序列与模板蛋白lajoA的氨基酸序列进行比对(如图1所示),一致性较好,达到75%,深蓝色表示氨基酸完全一致,代表一致性,浅蓝色表示氨基酸不一样但性质相近,代表相似性,序列比对结果比较理想,满足一致性大于60%的条件,因此选择lajoA为模板蛋白。以lajoA为模板进行同源建模后,可以看到建模后的3tyz三维结构(如图2a所示)。拉氏构象图中蓝线内为“最适区”,该区域的氨基酸数量越多,结构越可信,紫色线内为“允许区”,表示结构可接受,红色点表示构象不合理的氨基酸需要优化。评估结果显示,氨基酸处于合理区间的比例大于95%,符合要求。图2c为Profile-3D评估法中每个氨基酸的打分情况,氨基酸打分低于0的只有2个,大部分氨基酸打分较高,说明模型质量较好。通过评估,建立的3tyz模型符合要求。3tyz蛋白模型对接后与3tyz蛋白复合物结晶结构进行对比,模型与结晶结构相似性非常高(图3a,3b)。以存在晶体结构的3tyz进行同源建模的目的是为了模型能够与晶体结构进行比较,没有实际的应用意义,通常有了晶体结构就不需要再进行蛋白建模。建模模型与晶体结构相比较可以更加直观的展示同源建模技术。
2.2 受体-配体分子对接及相互作用关系
蛋白模型与配体SMZ能够很好地进行对接,对接结果显示,氨基酸THR(73)、ARG(74)和ARG(266)与SMZ发生氢键相互作用,用绿色表示;氨基酸LYS(232)与SMZ发生弱氢键相互作用,用浅绿色表示;氨基酸PHE(201)、PRO(75)、ARG(74)、LYS(232)与配体存在范德华力,较强的力用紫色表示,较弱的力用浅紫色表示(图4所示)。最优分子对接结果表明,受体3tyz与配体SMZ主要通过疏水作用力(图5a)、氢键(图5b)等作用力结合在一起。受体-配体间的范德华力一般较弱,起次要作用。这与通常认为的受体-配体相互作用规律一致,一般情况下,疏水作用力的影响最大,氢键次之[12]。
3 展望
在学习药物化学时正确、有效地使用DS软件,开展同源建模和分子对接研究,可让原本难以理解的受体-药物分子互作的微观过程以图形或动态方式呈现,使枯燥乏味的学习活动变得生动形象、易于接受,可大幅提高学习效率,为我国兽药研发领域培养合格的药学人才提供可靠而有力的技术支撑。
