敲除S1PR3可缓解小鼠的急性肺损伤:基于抑制MAPK信号通路
2022-02-03房尚萍孙任珂马同军
房尚萍,袁 冉,孙任珂,马同军
皖南医学院1麻醉学院,2麻醉学实验实训中心,3法医学院,安徽 芜湖 241002
脓毒症是病原微生物入侵机体后所致的全身炎症反应综合征[1,2]。肺脏是脓毒症中受累的首位靶器官[3]。在病程早期就可出现急性肺损伤,其发生率在危重病人群中高达7%。死亡率高达30%~40%,已成为脓毒症患者死亡的首要原因[4]。鞘氨醇1磷酸酯受体3(S1PR3)作为G蛋白偶联受体之一,与磷脂分子-1磷脂鞘氨醇结合,启动细胞跨膜信号转导,调节细胞增殖和炎症反应[5,6]。S1PR3在全身各组织器官内高表达[7]。有研究报道S1PR3是参与脓毒症过程巨噬细胞清除细菌的关键分子[8]。当干扰S1PR3表达可抑制LPS诱导的小胶质细胞TNF-α、IL-6和IL-1β表达[9]。研究显示S1PR3在急性肺损伤中是关键的分子[10]。
S1PR3 在脓毒性急性肺损伤的具体机制尚不明确。因此,本研究通过脂多糖(LPS)构建急性肺损伤模型,观察了敲除S1PR3和S1PR3激动剂对急性肺损伤的影响,及是否通过MAPK通路发挥作用,为急性肺损伤的治疗提供一定的实验依据。
1 材料和方法
1.1 材料
1.1.1 试剂 脂多糖(Sigma),S1PR3激动剂CYM-5541(MedChemExpress);caspase-1 抗体、clv-GSDMD 抗体,p-JNK抗体、p-ERK抗体、p-p38抗体(Cell Signaling Technology);ELISA试剂盒(武汉艾美捷科技有限公司)。
1.1.2 实验动物 健康SPF级雄性C57Bl/6J小鼠16只和S1PR3-/-敲除鼠16只,6~8周龄,体质量18~22 g,购自斯贝福(北京)生物技术有限公司,动物使用许可证号SCXK(京)2019-0010,本实验动物研究符合皖南医学院实验动物伦理委员会所制定的伦理学标准,动物伦理批准号为2021-201,实验遵循3R原则。
1.2 方法
1.2.1 急性肺损伤模型构建[11]所有小鼠经腹腔注射10%水合氯醛(0.20 mL/20 g)麻醉小鼠,固定于动物架上,置于工作台,抬高头端,使其与操作台面成30°角,用宽头镊拉出舌头,充分暴露声门,向气管内缓慢滴注LPS/NS(5 mg/kg),保持小鼠直立体位,直至完全吸入,恢复正常呼吸,造模后6 h,处理小鼠。
1.2.2 动物分组 雄性C57BL/6J 小鼠(6~8 周)分为2组:C57 NS组(C57,生理盐水处理)、C57 LPS组(C57,LPS诱导),8只/组;S1PR3敲除雄性小鼠(6~8周)分为2 组:S1PR3-/-NS 组(S1PR3 敲除鼠,生理盐水处理)、S1PR3-/-LPS组(S1PR3敲除鼠,LPS诱导)。
1.2.3 细胞分组 Ⅱ型肺泡上皮细胞(MLE-12细胞)铺板于6孔板中,每孔细胞数量为1×106,分为4组,每组3个副孔:PBS 组、LPS 组、CYM5541 组(仅加入CYM-5541)、CYM5541+LPS 组(加入CYM-5541+LPS),CYM-5541(10 nmol/L)预刺激2 h,加入LPS(1 μg/mL)刺激6 h。
1.2.4 HE染色 4%福尔马林固定肺组织,石蜡包埋,切成5 mm切片。然后,用HE染色切片。
1.2.5 Western blot 各组收集的肺组织或细胞,PBS清洗2次,加入RIPA试剂,蛋白酶抑制剂和磷酸酶抑制剂提取组织总蛋白,BCA法测量蛋白浓度,行SDS-PAGE电泳,转膜。5%脱脂奶粉封闭1 h,将条带分贝加入caspase-1,GSDMD,p-JNK,p-ERK p-p38 和内参抗体中,4 ℃避光孵育过夜,1×TBST洗膜3次,5 min/次,再置于二抗(1∶1000)孵育液中,室温孵育1 h,最后ECL显影液避光显影,使用Image J软件进行结果分析,通过目的蛋白灰度值与内参条带灰度值的比值表示目的蛋白的表达水平。
1.2.6 RT-qPCR 使用Trizol 法提取肺组织或细胞总RNA,逆转录合成cDNA,然后行PCR扩增。扩增条件:95 ℃3 min,95 ℃5 s,60 ℃30 s,40个循环。引物序列如下(表1)。
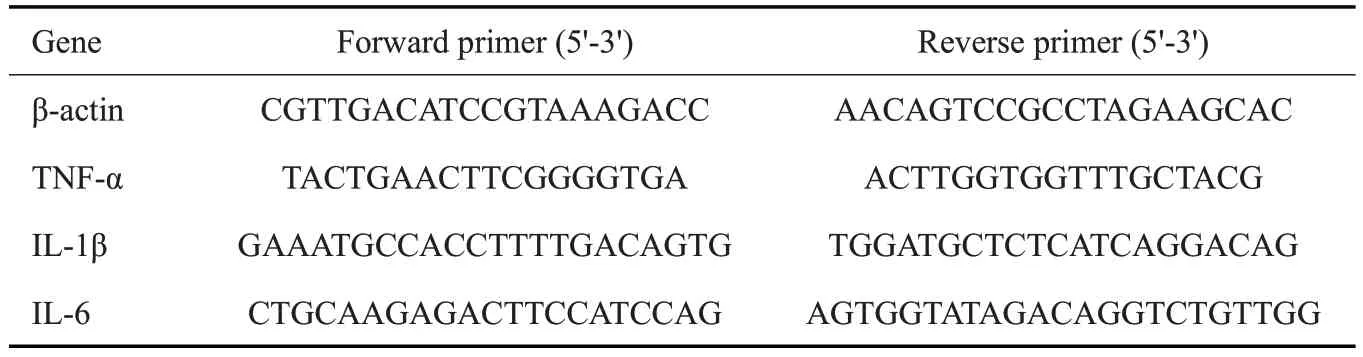
表1 qRT-PCR引物序列Table 1 Primer sequence for qRT-PCR
1.2.7 肺水肿检测 采用肺湿干比反应肺水肿情况。小鼠麻醉后右心灌流去除肺部血管内血液,取肺脏并称重,记作湿重。将肺脏至于65 ℃恒温烤箱,72 h再次称重,记作干重。
1.3 统计学分析
SPSS 22.0 软件进行实验数据分析。计量资料以均数±标准差表示。两组间比较用独立样本t检验;多组间比较用单因素方差分析,进一步两两比较用SNK-q检验。以P<0.05表示差异有统计学意义。所有的实验都是独立重复3次。
2 结果
2.1 LPS诱导的急性肺损伤中S1PR3表达上调
S1PR3在LPS处理的C57小鼠肺组织中表达明显上升(图1A,P<0.001),同时pro-IL-18,pro-IL-1β,TNF-α,IL-6 mRNA表达明显上升(图1B~C,P<0.05),小鼠血清IL-18,IL-1β较空白对照组均明显升高(图1D,P<0.05)。
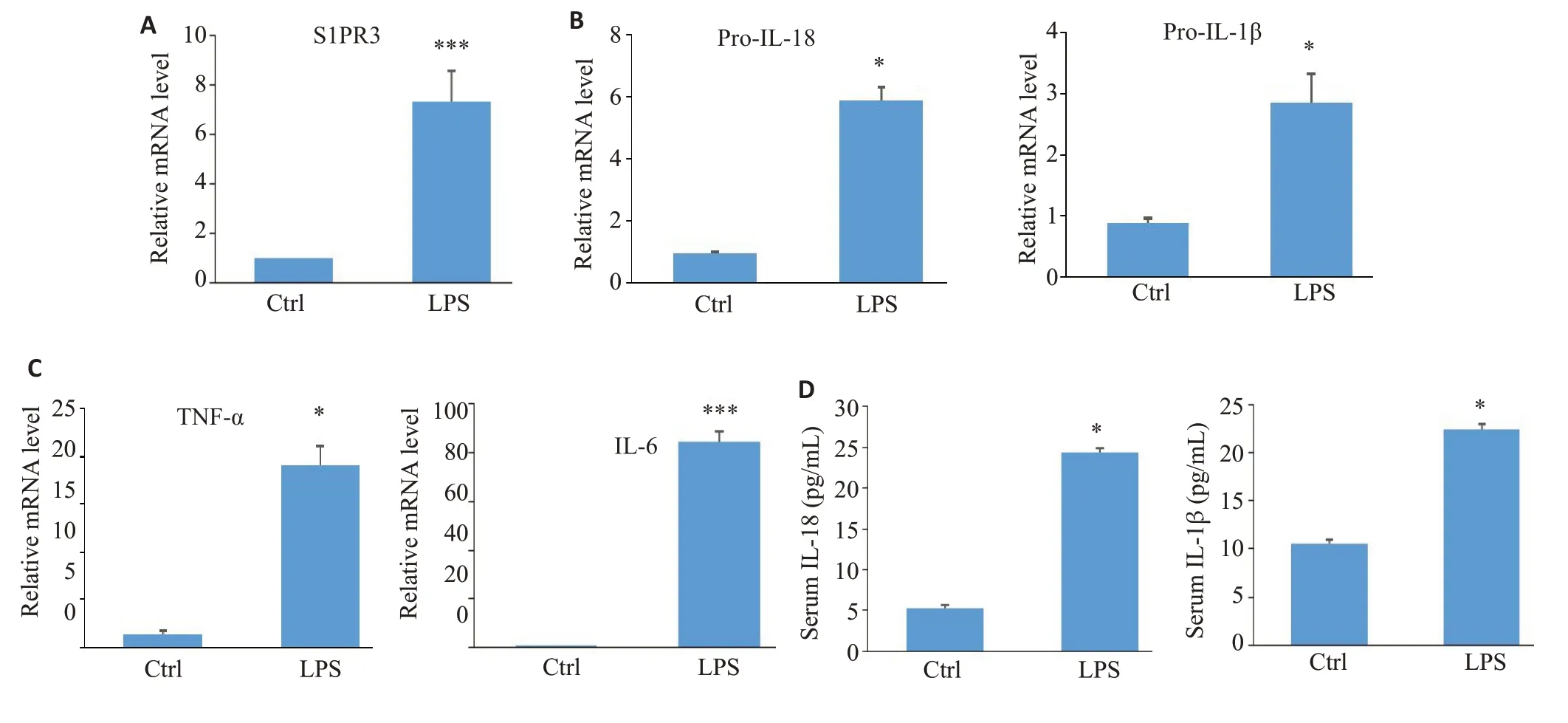
图1 急性肺损伤对S1PR3和炎症因子的影响Fig.1 Effect of acute lung injury on S1PR3 and inflammatory factors in C57 mice.A-C:qRT-PCR for detecting changes of S1PR3,pro-IL-18,pro-IL-1β,TNF-α,and IL-6 mRNAexpression in lung tissues.D:ELISAfor detecting changes of IL-18 and IL-1β expressions in serum.*P<0.05,***P<0.001 vs ctrl group
2.2 S1PR3敲除改善急性肺损伤出血
S1PR3-/-较WT小鼠经LPS刺激后的肺组织HE染色,肺损伤明显减轻(图2A~E)。同时WT和S1PR3-/-经LPS刺激后肺脏湿干比值均显著升高(图2F,P<0.05),而S1PR3-/-较WT小鼠经LPS刺激后的肺湿干比重明显降低。
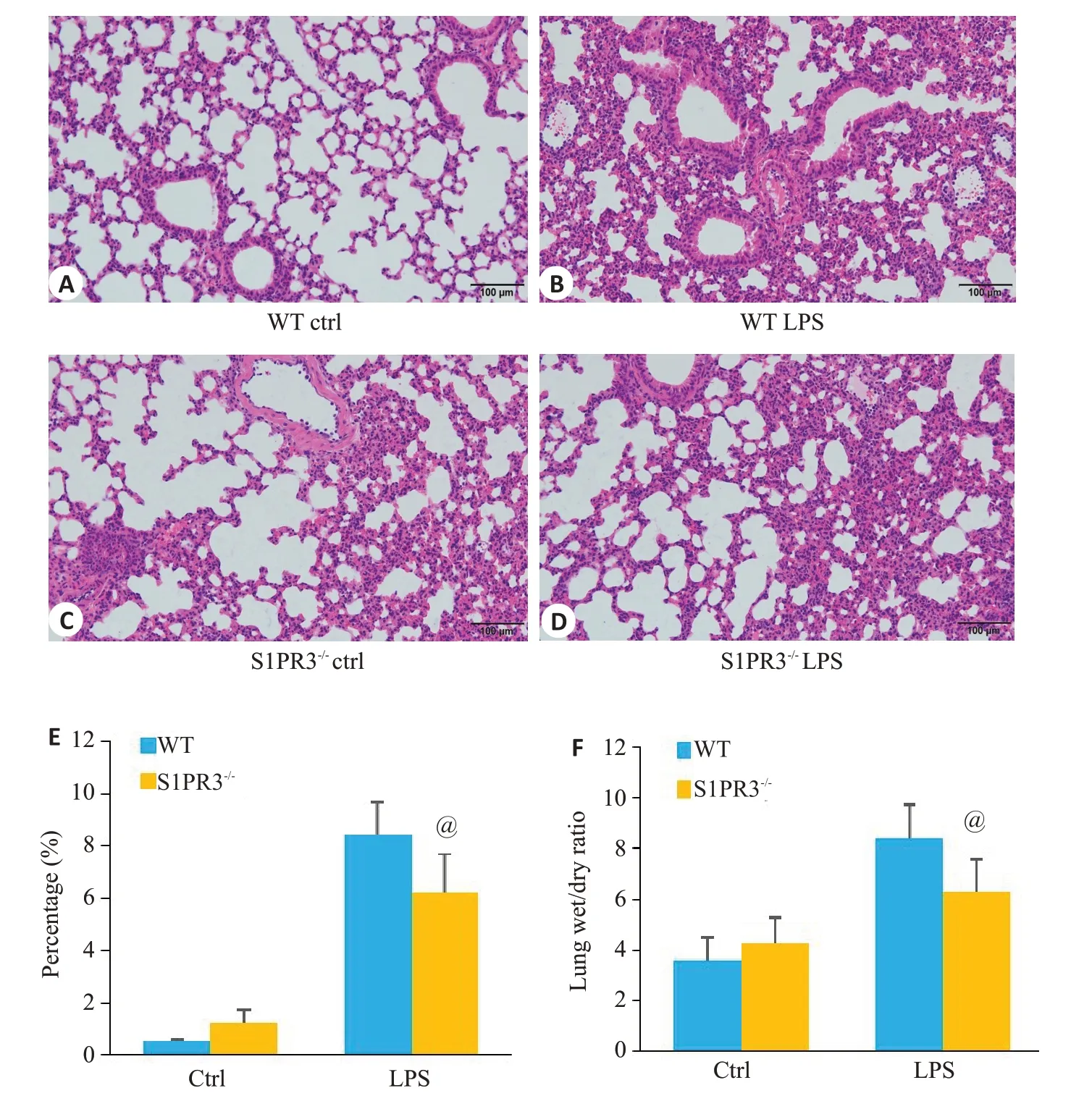
图2 S1PR3敲除对急性肺损伤病理的影响Fig.2 Effect of S1PR3 knockout on lung pathology in mice with acute lung injury.A-D:HE staining of the lung tissues.E: Percentage of neutrophil infiltration.F: Lung wet/dry ratio.@P<0.05 vs WT LPS Group.
2.3 S1PR3会加重细胞焦亡比例
S1PR3敲除后的小鼠急性肺损伤,血清乳酸脱氢酶(LDH)(图3A)和肺泡巨噬细胞的凋亡比例(图3B)均降低,cleaved-caspase-1和GSDMD蛋白表达(图3C~E)也较WT小鼠急性肺损伤时表达降低。II型肺泡上皮细胞因加入LPS 和S1PR3 激动剂后,细胞上清中的LDH 含量(图3I)明显升高,cleaved caspase-1 和GSDMD(图3F~H)蛋白表达也明显上升(P<0.05)。
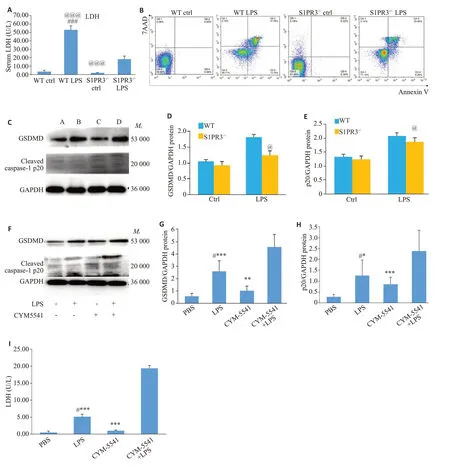
图3 S1PR3对急性肺损伤细胞焦亡的影响Fig.3 Pyroptosis in the lung tissue of S1PR3 knockout mice with acute lung injury and in MLE-12 cells treated with S1PR3 agonist.A: Changes of serum LDH levels in WT and S1PR3-/-mice.B:Flow cytometry for detecting pyroptosis of alveolar macrophages in mice with acute lung injury.C-E: Western blotting for detecting changes in GSDMD and cleaved caspase-1 p20 in the lungs of mice with acute lung injury(In Fig.3C,A,B,C and D represent WT Ctrl group,WT LPS group,S1PR3-/- Ctrl group,and S1PR3-/- LPS group,respectively).F-H:Western blotting for detecting changes in GSDMD and cleaved caspase-1 p20 in MLE-12 cells treated with CYM5541.I: Changes of LDH levels in MLE-12 cell supernatant.#P<0.05 vs PBS Group,@P<0.05 vs WT LPS Group,*P<0.05,***P<0.001 vs CYM5541+LPS group.
2.4 S1PR3敲除降低MAPKs通路的激活
急性肺损伤的小鼠肺组织中的p-JNK、p-ERK和p-p38表达均明显增强,而S1PR3敲除后减轻了这些分子的磷酸化(P<0.05,图4A~D)。而II型肺泡上皮细胞因加入LPS后MAPKs家族蛋白的磷酸化水平明显升高,而同时加入S1PR3激动剂和LPS后,MAPKs家族蛋白的磷酸化水平较单独加入LPS升高更明显(P<0.05,图4E~H)。
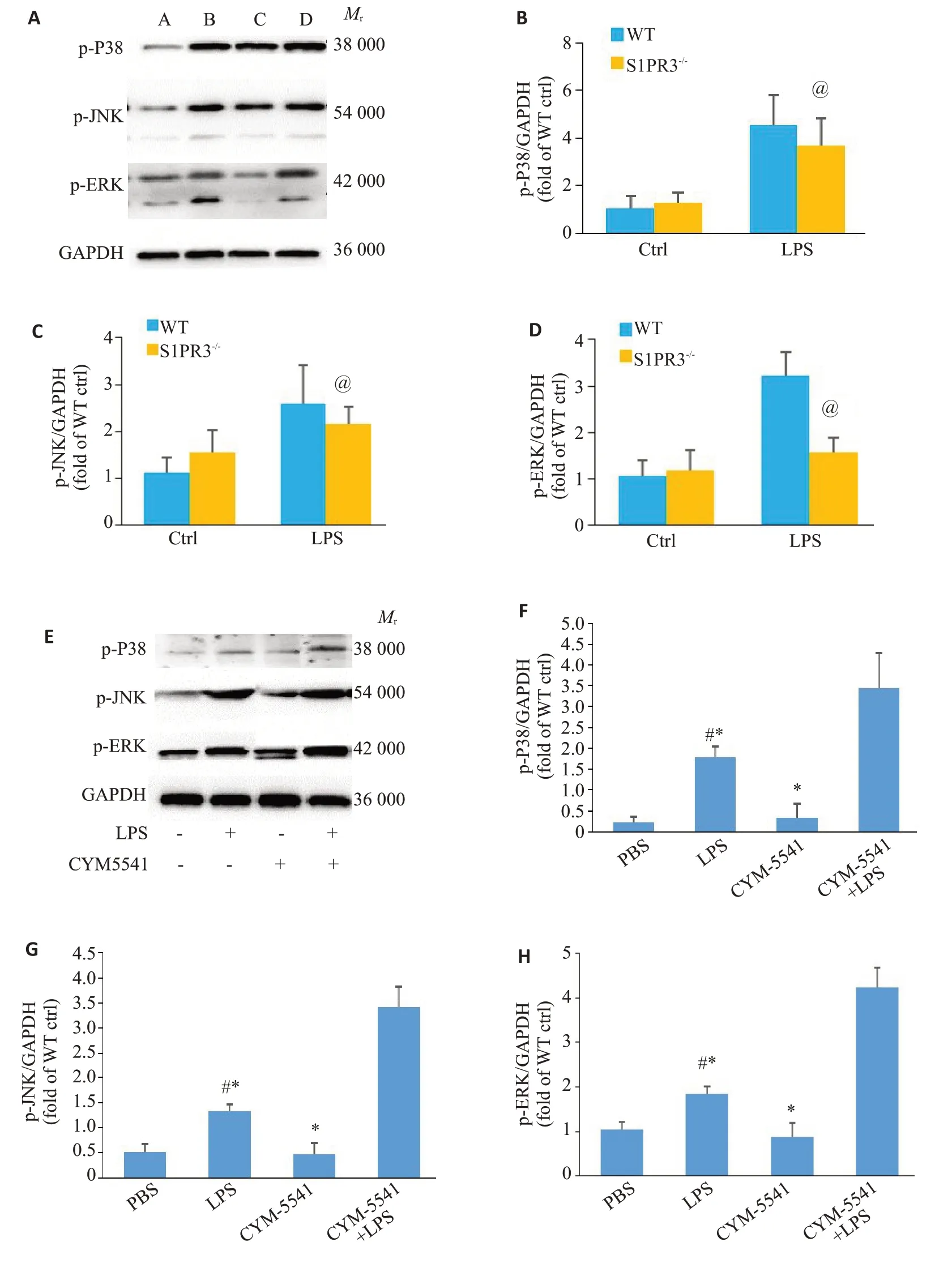
图4 S1PR3对MAPKs信号通路的影响Fig.4 Changes in MAPKs signal pathway in the lung tissue of S1PR3 knockout mice with acute lung injury and in MLE-12 cells treated with S1PR3 agonist.A-D:Western blotting for detecting changes in p-p38,p-JNK and p-ERK in the lungs of the mice with acute lung injury(in Fig.4A,A,B,C and D represent WT Ctrl group,WT LPS group,S1PR3-/-Ctrl group,and S1PR3-/-LPS group,respectively).E-H:Western blotting for detecting changes in p-p38,p-JNK and p-ERK in MLE-12 cells treated with CYM5541.#P<0.05 vs PBS Group,@P<0.05 vs WT LPS Group,*P<0.05 vs CYM5541+LPS group.
3 讨论
本研究首次发现S1PR3促进急性肺损伤发展,通过caspase-1经典的细胞焦亡途径,通过抑制S1PR3表达会抑制caspase-1细胞焦亡途径,抑制急性肺损伤的发展;反之,过表达S1PR3促进caspase-1细胞焦亡途径,促进急性肺损伤的发展。
脂多糖作为革兰阴性杆菌细胞壁外膜内毒素的主要成分,是引起脓毒症和急性肺损伤的主要致病因素[12,13]。本研究中,向气管内注入LPS直接诱导急性肺损伤,病理结果发现LPS滴注气管后导致大量炎症细胞浸入肺泡腔和肺毛细血管充血,肺泡壁增厚;同时,血清炎症因子TNF-α,IL-6,IL-1β明显升高,说明急性肺损伤的模型构建成功,该结果与某报道一致[14]。S1PR3广泛分布于各类组织细胞中,是七次跨膜G蛋白偶联受体家族的其中一员,与磷脂分子1-磷酸鞘氨醇(S1P)相结合,启动下游信号转导,可调节细胞增殖,炎症等生物学功能[10,14]。本研究发现急性肺损伤后S1PR3表达明显升高,该结果与某研究结果类似:急性肺损伤小鼠血浆中S1PR3明显上升[10]。出现该结果的可能机制是S1PR3通过介导细胞增殖和增强血管通透性。而在S1PR3-/-小鼠急性肺损伤中,肺组织渗出的炎症细胞和毛细血管充血程度明显减轻。研究发现S1PR3拮抗剂能抑制小胶质细胞炎症因子的产生,从而抑制脓毒性脑病的发生发展[16],该现象与本结果类似。说明S1PR3可促进脓毒症的发生发展,而敲除S1PR3能明显缓解脓毒症的进程。
本研究发现S1PR3-/-急性肺损伤小鼠,肺组织细胞焦亡比例明显减少,分泌的LDH明显降低,炎症反应明显减轻。与本研究结果类似的研究显示S1PR2缺失的急性肺损伤巨噬细胞焦亡减少,脓毒症的发生明显减轻[17]。S1PR2和S1PR3均是S1PRs家族中的主要成员,高度同源,具有相似的生物学效应[18,19]。但S1PR3缺失是否通过减轻细胞焦亡,而减缓脓毒症的发生鲜有报道,这也是本研究的创新点之一。细胞焦亡是一种促炎性的细胞死亡方式,细胞在感受到外来信号刺激后,激活炎性小体[20],caspase-1前体蛋白被剪切,形成具有活性的caspase-1,同时剪切IL-1β和IL-18前体形成具有生物活性的成熟体[20,21]。同时Caspase-1通过剪切gasdermin D(GSDMD)蛋白诱导细胞焦亡,细胞肿胀发生渗透性溶解[22]。本研究发现S1PR3-/-小鼠脓毒性肺损伤后,肺组织GSDMD及clv-caspase-1等焦亡相关蛋白明显减少。II型肺泡上皮细胞是肺泡上皮细胞的主要组成细胞,是参与急性肺损伤的重要效应细胞[23],因此本实验采用II型肺泡上皮细胞系MLE-12细胞模拟体外实验。CYM5541 是选择性S1P 受体3 的变构激动剂,能模拟S1PR3过表达后的生物学效应[24]。本研究发现MLE-12细胞加入LPS和CYM5541后,细胞焦亡相关蛋白表达明显升高,与S1PR3敲除后现象相反。说明S1PR3会促进细胞焦亡的发生。
MAPKs家族是介导细胞内信号传导的一组丝氨酸/苏氨酸激酶,该通路被激活后会导致细胞增殖、分化、死亡和促炎症介质的产生等效应[25,26]。MAPKs通路也被认为是急性肺损伤的关键信号[27,28]。目前鲜有研究报道MAPKs家族在S1PR3介导的急性肺损伤的发展中起作用。Tian等发现JNK、ERK和p38在急性脓毒症肝损伤模型中被活化,而MAPKs通路会被S1PR3抑制剂所抑制[29],该研究与本研究结果类似,JNK、ERK和p38MAPKs家族在急性肺损伤时被激活,而S1PR3敲除小鼠的急性肺损伤中MAPK通路被抑制。S1PR3激动剂会使MAPKs通路相关蛋白的磷酸化水平升高。
综上所述,敲除S1PR3通过抑制MAPKs信号通路抑制细胞焦亡,从而改善脓毒性急性肺损伤。
