Kartagener 综合征1例报告
2022-01-28韩晓博张宇涵张信信孙天宇王韧韬解立新
韩晓博,张宇涵,张信信,孙天宇,王韧韬,解立新
1 解放军总医院第八医学中心 呼吸与危重症学部,北京 100091;2 解放军医学院,北京 100853
Kartagener 综合征是以慢性鼻窦炎、支气管扩张和内脏转位三联征为临床特征的一种罕见遗传病,是由于基因突变引起的纤毛结构和(或)功能缺陷导致了一系列与纤毛运动障碍有关的临床症状[1]。在胚胎发育早期负责控制心脏和内脏器官正常位置的胚节纤毛发生功能障碍,可以使内脏随机转位。在出生后由于纤毛的清除黏液功能,患者可出现反复的下呼吸道感染、中耳炎、鼻窦炎等。精子纤毛功能不良可导致男性不育,输卵管纤毛异常可延迟卵子运输导致女性不孕[2]。Kartagener 综合征患病率为1/40 000~ 1/20 000[3],通常的遗传方式是常染色体隐性遗传,也有少数X 染色体遗传的报道[4-5]。由于该病患病率低,临床表现复杂,缺乏早期诊断的方法,容易出现漏诊及延误诊治的情况[6]。近年来外显子基因测序成为早期筛查和识别原发纤毛不动的重要手段,目前仍有未知的突变基因需要继续探索。因此本文通过回顾1例完全型Kartagener 综合征患者11年随诊临床资料,结合文献复习总结原发性纤毛运动障碍(primary ciliary dyskinesia,PCD)诊断方法的研究进展,探讨全外显子基因检测对早期诊断PCD 的价值。
病例资料
1 病史 患者,女性,40 岁。因反复咳嗽咳痰16年,加重1个月于2020年12 月20 日入解放军总医院第八医学中心。患者自2004年开始无诱因出现反复咳嗽,咳黄痰,间断应用抗生素及化痰平喘药物治疗可缓解。2009年因咳嗽加重至解放军总医院第八医学中心就诊,肺CT 提示内脏转位,支气管扩张伴感染,考虑Kartagener 综合征可能。2016年开始出现活动后喘憋,运动耐力下降。因活动后憋气症状加重1个月于解放军总医院第八医学中心住院治疗。既往10年鼻窦炎及中耳炎病史,自幼丧失嗅觉。家族无其他人有慢性呼吸道感染病史。病历资料已获得患者知情同意。
2 查体 体温36.5℃,脉搏80/min,呼吸频率20 次/min,血压120/78 mmHg(1 mmHg=0.133 kPa);口唇无发绀,右侧鼻腔可见半透明新生物,粗侧听力正常。颈软,无颈静脉怒张;听诊双肺可闻及痰鸣音;心脏位于右侧,右侧锁骨中线第5 肋间隙内0.5 cm 处可触及心尖冲动。腹软,无压痛,四肢活动好。
3 辅助检查:经鼻呼气一氧化氮:8 bbp。肺功能提示中度阻塞性通气功能障碍,呼气峰流速下降,弥散功能正常。肺CT:内脏完全转位,双肺弥漫分布微结节,多发囊状扩张支气管,双肺下叶支气管管腔内可见高密度影填充,左肺中叶体积缩小,呈三角形高密度影,2009年与2020年肺CT 比较可见支气管扩张明显加重(图1)。副鼻窦CT 提示双侧上颌窦、筛窦、额窦、蝶窦内见密度增高影,慢性鼻炎(图2C)。鼻内镜可见右侧中鼻道息肉组织。支气管镜检查可见主气道大量黏性分泌物,取支气管黏膜组织刷检送电镜检测可见部分纤毛外动力臂缺失及复合纤毛(图3)。结合患者病史、临床症状体征、电镜检查结果,确诊为Kartagener 综合征(完全型Kartagener 综合征影像检查见图3)。
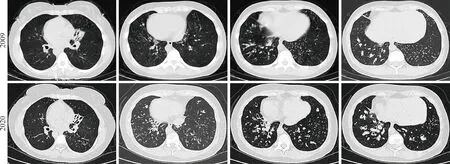
图1 2009年和2020年肺部CT 进展Fig.1 Evolution of lung abnormalities on chest CT in 2009 and 2020
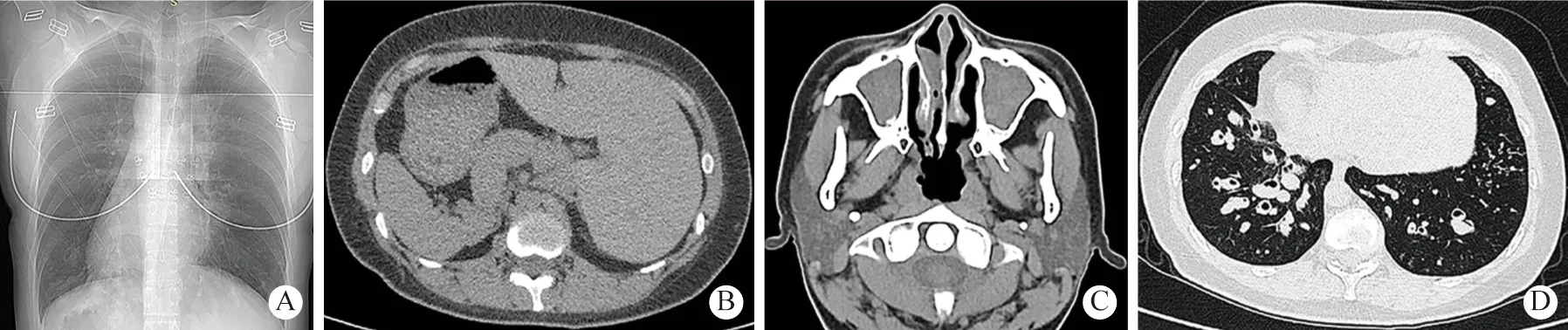
图2 Kartagener 综合征影像图片A 和B:内脏转位(右位心,肝在左,脾在右);C:多组鼻窦炎,鼻炎;D:双下肺囊性支气管扩张及树芽征Fig.2 Radiographic images of Kartagener syndromeA and B:situs inversus totalis;C:sinusitis and rhinitis;D:cystic bronchiectasis and tree-in-bud in the lower lung

图3 支气管黏膜电镜结果:A 部分纤毛外动力蛋白臂缺失,B 部分纤毛外动力蛋白臂缺失及复合纤毛Fig.3 Findings under transmission electron microscope (TEM):ultrastructural ciliary defects were observed such as partial outer dynein arm defect (A) and compound cilia (B)
4 外显子基因检测:采集全血标本,提取基因组DNA:采用The Sure Select Human All Exon V6 定制芯片捕获外显子,Illumina 测序平台进行高通量测序对疑似致病突变的目标序列进行PCR 后,经ABI373 进行Sanger 测序验证,测序结果经DNASTAR 子程序SeqMan 软件得到验证。结果患者于DHAH5基因上有一个杂合突变(图4),DHAH5基因63 号外显子出现一个杂合突变,在10616 核苷酸由鸟嘌呤G 变为腺嘌呤A(c.G10616A) 的突变(箭头所示),导致第3539 号氨基酸由精氨酸变为组氨酸(p.R3539H)。该位点为国外已报道位点[14]。
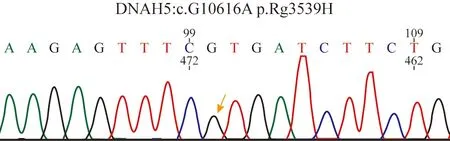
图4 全外显子基因检测:DNAH5 基因63 号外显子c.G10616A突变Fig.4 Mon heterozygous mutation (c.G10616A) in exon 63 of the DNAH5 gene
5 治疗 患者口服头孢克肟(0.2 g,2 次/d)及左氧氟沙星(0.6 g,1 次/d)抗感染治疗,并联合乙酰半胱氨酸溶液(3 mL,2 次/d) 化痰治疗后好转,于2020年12 月24 日出院。出院后长期门诊随诊指导患者体液引流排痰及肺康复锻炼。
讨 论
Kartagener 综合征属于PCD 的一种亚型,国外研究发现Kartagener 综合征约占PCD 的50%,推测在胚胎发育中由于胚节纤毛功能障碍,内脏出现转位或不转位的随机事件[7-8]。Kartagener 综合征并不会在出生时即会表现所有临床症状,随着年龄增长,患者复发慢性咳嗽和呼吸道感染,临床症状逐渐典型。对于表现为完全型Kartagener综合征的患者(同时出现鼻窦炎、支气管扩张、内脏转位三联征),诊断相对容易,但确诊时间往往延迟。而不伴有内脏转位的PCD 临床异质性较大,加之临床医生认识水平不足,极易出现漏诊和诊断延误[9]。本例Kartagener 综合征患者在年幼时常出现反复咳嗽咳痰症状,直至中年确诊。我们对比了患者前后11年随访的肺CT 变化,可见肺部病变主要集中在双下肺,支气管扩张并感染逐渐加重(图1)。本研究首次对比了Kartagener 综合征确诊前后11年肺CT 的变化,可以更直观地认识疾病的进展,目前国内缺乏Kartagener 综合征长期随访的肺功能及影像学数据,很多患者确诊时已进展至终末状态,早期确诊PCD,尽早开始健康指导及肺康复锻炼对于改善患者预后极为重要。
对于临床疑似PCD 的患者,目前常用检测方法主要有鼻一氧化氮测定、纤毛透射电镜分析(transmission electron microscopy,TEM)、高速视频显微镜分析(high speed video-microscopy analysis,HSVA)、基因分析和免疫荧光检测[10-11]。TEM 是既往确诊PCD 的重要方法,但仍有30%的患者纤毛电镜下未见超微结构异常[12]。随着基因检测技术发展,外显子基因检测技术已作为重要的诊断技术用于临床。鉴于检测技术的局限性,没有单一的检测试验可作为诊断PCD 参考的“金标准”。2018年ATS 指南对PCD 的诊治流程给出建议,将满足以下4 条临床症状中2 条及以上的患者作为PCD 筛查的高危人群:1)足月儿出现原因不明的呼吸窘迫;2)6 月龄内起病的常年咳嗽;3)6 月龄以内起病的常年鼻塞;4)内脏转位。对高危人群建议采用鼻一氧化氮测定做初步筛查,疑似病例采用扩充基因检测发现PCD 相关基因的双等位基因变异明确诊断,无明确基因变异的高度疑似患者需进一步寻找电镜下纤毛结构异常和纤毛活动异常的证据。临床症状综合上述检测方法可以提高疾病的诊断能力。本例患者符合完全型Kartagener 综合征的临床表现,透射电镜可见部分纤毛外动力臂缺失及复合纤毛,支持了原发性纤毛功能障碍的确诊。
原发性纤毛运动障碍的致病基因复杂多样,近年来与纤毛不动相关的新基因不断被发现,目前已报道了40 种可导致PCD 的基因变异[4,13]。2018年ATS 指南及欧洲呼吸学会发表的指南均支持早期基因检测作为PCD 的一项确诊依据[12,14]。常见基因变异包括编码轴丝外动力蛋白臂的DNAH5、DNAH9、DNAH12、DNAI1、ARMC4、CCDC103 以及内动力蛋白臂的DNALI1 等[15]。不同的突变基因可能与疾病表型和预后相关[16]。本例Kartagener 综合征患者的全外显子测序仅检测到DNAH5 基因一个变异位点,与既往报道的纯合或复合杂合的双等位基因突变遗传方式不相符[4]。研究发现DNAH5 是PCD 的高频致病基因,目前报道的PCD 基因检测中DNAH5 占28%~ 35%[17]。PCD 是否存在单基因杂合突变致病仍不清楚。一项在对意大利47例PCD 患者的研究中可见6例患者存在单基因杂合突变,其中4例为DNAH5 单基因杂合突变[18]。我国广州呼吸病研究所报道的5例Kartagener 综合征病例系列研究中也发现有2例患者为单杂合突变[19]。目前仍有约30%的PCD 患者无法通过基因检测确定诊断,因此基因检测阴性并不能排除PCD 的诊断。对于本例PCD患者出现单杂合基因突变,可能的解释包括:受限于外显子基因检测技术,编码区的下一代测序筛查存在无法检测到的致病突变(如启动子、内含子和其他调控序列的突变)[18];PCD 基因之间的相互作用,以及存在未知突变基因的可能[20];本例患者携带有单基因杂合DNAH5 突变,但不能确定是否为单基因杂合DNAH5 突变致病。
总之,PCD 是临床异质性及基因异质性均较强的罕见遗传病,漏诊和延误诊断相当普遍。我们在国内首次报道了Kartagener 综合征随诊11年肺部影像学变化,以加深对疾病进展的认识。随着基因检测技术的快速发展,全外显子基因检测技术已应用于临床,但仍有很多未知的突变基因和基因之间相互作用有待探索。随着病例和基因数据的积累及PCD 基因突变数据库的建立,基因检测技术对PCD 的早期诊断能力将逐渐提高。对确诊患者及家族成员进行基因检测,也有助于早期诊断家族中其他PCD 成员,并为产前筛查和遗传咨询提供依据。
