超高效液相色谱-四极杆/静电场轨道阱质谱测定猪尿中环丙氨嗪及其代谢物残留量
2022-01-09罗秋红邓智伟万建春占春瑞
罗秋红,韩 颖,胡 蓓,邓智伟,曾 臻,万建春,占春瑞
(南昌海关技术中心,江西 南昌 330038)
环丙氨嗪(Cyromazine)是一种含氮杂环类杀虫剂,2002 年被农业部批准为三类新兽药。在养殖场中,环丙氨嗪主要作为预混剂拌入饲料,通过饲喂的方式进入动物体内,其中大部分以原型及其代谢物的形式随尿液和粪便排出体外,沉积在粪便中的环丙氨嗪可杀灭蝇蛆[1]。因作用效果明显,导致养殖户将环丙氨嗪长期应用于猪饲料中而没有休药期,造成了动物性产品的药物残留。虽然研究表明环丙氨嗪的毒性较小,但其代谢产物三聚氰胺(Melamine)能导致小鼠膀胱肿瘤,是一种潜在的致癌物质,因此我国制定了动物源食品中环丙氨嗪的残留限量[2]。三聚氰胺可与三聚氰酸形成不溶性络合物,导致猫急性肾功能衰竭而死亡[3]。目前动物源食品中环内氨嗪和三聚氰胺的检测较受关注,也出台了相应的检测方法标准,但对于饲养环节控制环丙氨嗪喂食量,监控尿样残留量与食用动物组织残留量的关联性研究未见报道。
目前,蔬菜、牛奶、肉制品等样本中环丙氨嗪和三聚氰胺的测定方法已有报道,包括酶联免疫法[4]、快速检测试纸条法[5]、高效液相色谱法[6-8]、液相色谱-串联质谱法[9]和毛细管电泳法[10]等。酶联免疫法和快速检测试纸条法的检测原理均为抗原抗体结合反应,检测速度快,但存在假阳性的可能。高效液相色谱法灵敏度较低,无法满足低含量样本的检测要求。毛细管电泳法在检测复杂样品时,杂质干扰较大。液相色谱-串联质谱法的抗基质干扰能力及灵敏度较高,但多反应监测模式(MRM)的定性能力有一定局限性。液相色谱-串联高分辨质谱技术具有灵敏度高、定性准确的特点,在农兽药等化学污染物的检测方面应用广泛[11-14],但关于猪尿中环丙氨嗪及其代谢物三聚氰胺残留的检测方法报道较少。
本文利用超高效液相色谱-四极杆/静电场轨道阱质谱(UHPLC-Quadrupole-Orbitrap MS),采用平行反应监测模式(PRM),建立了猪尿中环丙氨嗪及其代谢物三聚氰胺残留量的检测方法,可为研究控制环丙氨嗪喂食量提供有效的检测手段。
1 实验部分
1.1 仪器与试剂
Q Exactive 质谱仪,配UltiMate 3000 超高效液相色谱仪(美国Thermo Fisher 公司);Agilent 6495 液相色谱-串联质谱仪(美国Agilent公司);N-EVAP氮吹仪(美国Organomation 公司);MCX固相萃取柱(60 mg/3mL,上海安谱公司);Milli-Q超纯水机(美国Millipore公司)。
环丙氨嗪(纯度99.26%,Dr.Ehrenstorfer 公司);三聚氰胺(纯度99.9%,TMstandard 公司);三聚氰胺-15N3(纯度98.8%,TRC公司);乙酸、氨水(分析纯,西陇科学股份有限公司);甲酸(色谱纯,科密欧化学试剂有限公司);乙酸铵(色谱纯,ACS公司);β-葡萄糖醛苷酸酶/芳基硫酸酯酶(>100 000 U/mL,上海安谱公司);三氯乙酸(色谱纯,西亚化学科技有限公司);甲醇、乙腈(色谱纯,默克公司);实验用水为Milli-Q超纯水。
1.2 标准溶液的配制
精确称取适量的环丙氨嗪、三聚氰胺以及三聚氰胺-15N3(内标),分别置于10 mL 容量瓶中,用95%乙腈水溶液溶解定容,配制质量浓度为1 mg/mL的标准储备溶液。取标准储备溶液,用95%乙腈水溶液配制质量浓度为100 ng/mL 的环丙氨嗪和三聚氰胺混合标准工作液以及1.0µg/mL 的内标工作液,于4 ℃下保存。
分别准确吸取混合标准工作液及内标工作液适量,用95%乙腈水溶液配制成质量浓度为0.2、1.0、5.0、10.0、20.0、50.0 ng/mL的系列标准溶液,其中内标质量浓度为10.0 ng/mL。
1.3 样品前处理
准确称取2.0 g 猪尿样本于离心管中,加入100 µL 内标工作液和6 mL 1%三氯乙酸溶液,涡旋1 min,超声5 min。以4 500 r/min 离心5 min,上清液转入经5 mL 甲醇和5 mL 水活化后的MCX 固相萃取柱中,用5 mL 2%甲酸水淋洗后,负压抽干MCX 柱,加入5 mL 5%的氨水甲醇洗脱,收集全部洗脱液。氮吹至干,加入1 mL 95%乙腈水溶液复溶,过0.22µm 微孔滤膜,样品溶液用95%乙腈水溶液稀释10倍后,待检测。
1.4 色谱条件
色谱柱为Syncronis HILIC 色谱柱(100 mm×2.1 mm,1.7µm),柱温为35 ℃;流动相:A 为乙腈,B 为10 mmol/L 乙酸铵水溶液。梯度洗脱程序:0~1 min,98% A;1~5 min,98%~80% A;5~5.5 min,80%~5%A;5.5~6.5 min,5%A;6.5~7.0 min,5%~98%A;7.0~10.0 min,98%A;流速:0.4 mL/min;进样量:2µL。
1.5 质谱条件
Q Exactive 质谱仪参数:离子源为可加热电喷雾离子源(HESI-Ⅱ);正离子扫描模式;离子传输管温度为320 ℃;喷雾电压为3.5 kV,鞘气流速为40 arb;辅助气流速为10 arb;PRM 模式;分辨率采用17 500;进入C 形阱的目标离子数(AGC target):2 × 105;监测离子对:三聚氰胺m/z127.072 5 >85.051 2(定量离子)、127.072 5>68.024 9;内标m/z130.063 2>87.045 0;环丙氨嗪m/z167.103 4>85.051 1(定量离子)、167.103 4>125.081 9;碰撞能量(CE)均为75%。
Agilent 6495液相色谱-串联质谱仪参数:电喷雾离子源;正离子扫描模式;毛细管电压为4 000 V;鞘气温度为350 ℃,鞘气流速为11 L/min;干燥气温度为150 ℃,干燥气流速为15 L/min;MRM 模式;监测离子对:三聚氰胺m/z127.0 > 85.0(CE:27 eV,定量离子)、127.0 > 68.2(CE:35 eV);内标m/z130.1 > 87.4(CE:30 eV);环丙氨嗪m/z167.0 > 85.0(CE:23 eV,定量离子)、167.0 > 125.2(CE:23 eV)。
2 结果与讨论
2.1 色谱条件的优化
实验比较了Hypersil GOLD C18(100 mm × 2.1 mm,1.7 µm)和Syncronis HILIC(100 mm × 2.1 mm,1.7µm)2 种色谱柱对环丙氨嗪和三聚氰胺的色谱保留能力。结果表明,环丙氨嗪和三聚氰胺在C18柱上的保留较差,基质共流出,杂质干扰较严重;在HILIC 色谱柱上的出峰时间分别为3.1 min 和4.8 min 左右,色谱保留能力较C18柱强,能减少强极性杂质的干扰,更适合环丙氨嗪和三聚氰胺的分离。分别考察了不同浓度(5、10、15、20 mmol/L)乙酸铵和乙腈作为流动相的分离效果。结果表明:乙酸铵浓度越低,三聚氰胺的响应越高;而环丙氨嗪在乙酸铵浓度为10 mmol/L 和15 mmol/L 时的响应较高。此外,乙酸铵浓度为10、15、20 mmol/L时,目标化合物的峰形均较好。由于流动相中盐浓度越高,对仪器损伤越大。综合考虑,实验选择10 mmol/L乙酸铵水溶液-乙腈为流动相。三聚氰胺及其内标、环丙氨嗪的提取离子流色谱图如图1所示。
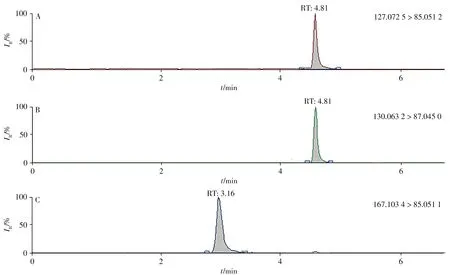
图1 UHPLC-Quadrupole-Orbitrap质谱上三聚氰胺(A)、内标(B)、环丙氨嗪(C)的提取离子流色谱图Fig.1 Extracted ion chromatograms of melamine(A),melamine-15N3(B)and cyromazine(C)on UHPLC-Quadrupole-Orbitrap MS
2.2 质谱参数的优化
质谱的碰撞能量对目标物的裂解有重要影响。UHPLC-Quadrupole-Orbitrap高分辨质谱的PRM与三重四极杆质谱的MRM模式相似,在Q1中分离出前体离子,然后并行记录所有生成的MS/MS碎片离子,最后以母离子>子离子进行定量,具有扫描全、质量准确、分辨率高等特点。配制1 mg/L的混合标准溶液,分别在MRM模式和PRM模式下对二级质谱的碰撞能量进行优化。在MRM模式下,根据碎片离子的相对响应强度,确定目标物合适的二级碰撞能量。在PRM模式下,根据碎片离子的相对响应强度和二级质谱信息确定合适的碰撞能量,使获得的谱图既有母离子的精确质量数,又有二级质谱的全扫描信息,满足定性和定量的要求。图2为UHPLC-Quadrupole-Orbitrap MS采集的二级质谱图,该图谱包含了未被碎裂的母离子精确质量数(m/z127.072 5、130.063 2和167.103 4)与所有二级碎片离子的精确质量数和丰度比。
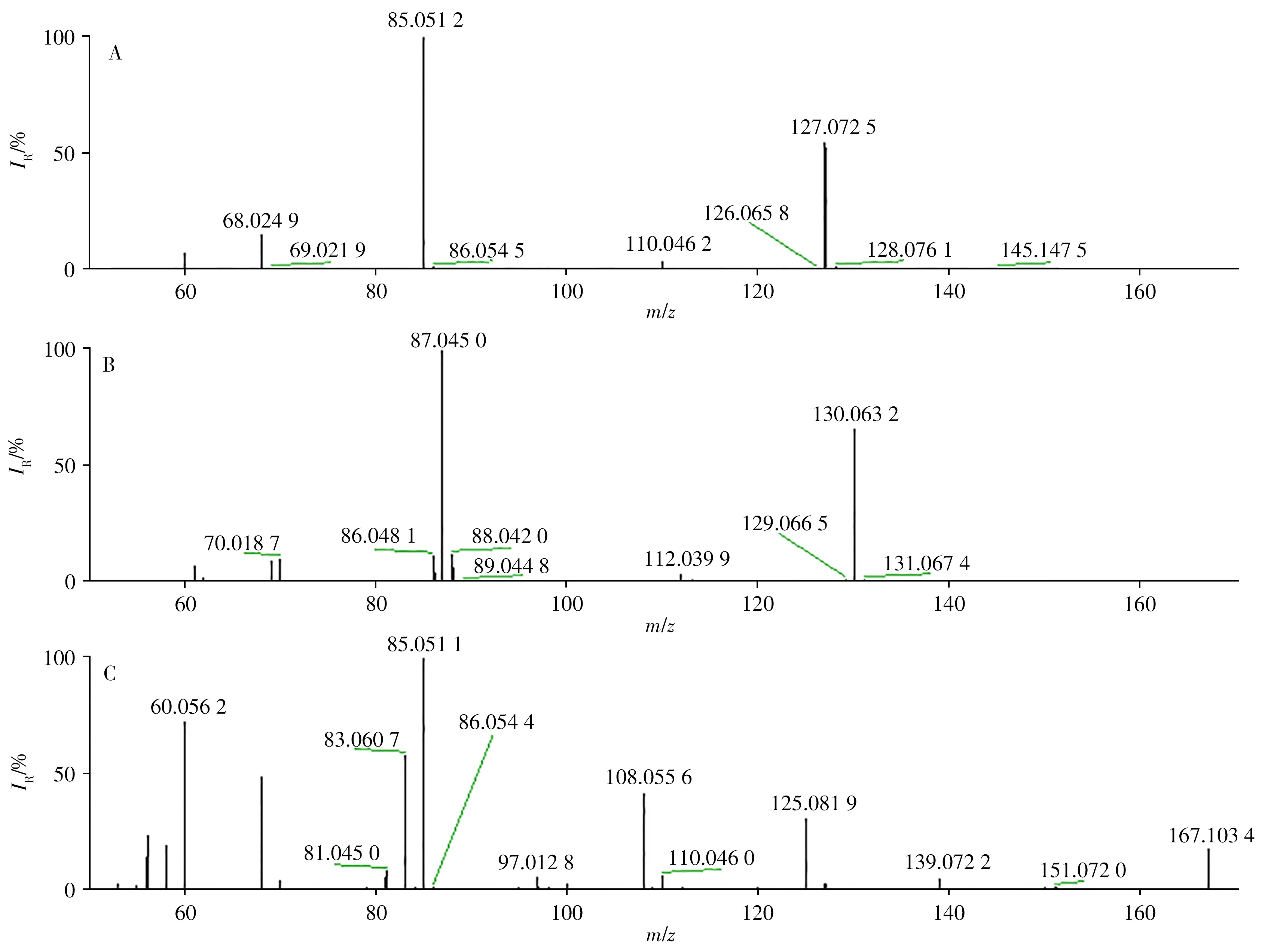
图2 UHPLC-Quadrupole-Orbitrap质谱上三聚氰胺(A)、内标(B)以及环丙氨嗪(C)的二级质谱图Fig.2 MS/MS spectra of melamine(A),melamine-15N3(B)and cyromazine(C)on UHPLC-Quadrupole-Orbitrap MS
欧盟委员会决议规定[15]:当采用普通低分辨质谱进行分析时,低分辨质谱-多级质谱的母离子可获得1个鉴定位点,多级质谱的产物离子可获得1.5个鉴定位点。质谱方法确证化合物需有4个鉴定位点,因此在确定母离子的基础上至少需要2 个子离子。低分辨质谱法[16]通常将碎片离子(m/z127.0 >68.2,167.0>125.2)作为辅助定性离子。而高分辨质谱-多级质谱的母离子为2个鉴定位点,多级质谱的产物离子为2.5个鉴定位点,本方法采集2个特征离子,鉴定位点数达到7。相比之下,高分辨质谱法的鉴定位点数更多,弥补了低分辨质谱易出现假阳性的缺点,具有较强的定性优势。
2.3 酶解试验
葡萄糖醛酸结合是药物代谢的重要结合方式之一。在葡萄糖醛酸转移酶的催化下,葡萄糖醛酸通过与底物的O、S、N 或C 原子连接形成轭合的代谢物,结合后其毒性降低,且易排出体外[17]。环丙氨嗪和三聚氰胺结构中均含有氨基,经过猪体内代谢,可能会产生葡萄醛糖酸结合物,因此对阳性猪尿样品进行酶解试验。结果发现,未酶解样品和采用β-葡萄糖醛苷酸酶/芳基硫酸酯酶酶解的样品中环丙氨嗪以及三聚氰胺的含量相差在15%以内,表明结合率比较低,两者主要以游离的形式存在,因此本实验采用不酶解的方式进行处理。
2.4 提取条件的优化
本研究对比了不同体积分数(50%、67%、100%)1%三氯乙酸乙腈溶液的提取效果,结果显示3种溶液对环丙氨嗪和三聚氰胺的提取效果差异不明显。因环丙氨嗪和三聚氰胺是极性化合物,同时为减少有机试剂的使用,本实验采用6 mL 1%三氯乙酸作为提取溶剂。经试验验证,猪尿样品中加入6 mL 1%三氯乙酸后,pH<3.0,满足MCX固相萃取柱的净化条件。
2.5 基质效应
分别采用试剂和基质样品建立标准曲线评估样品的基质效应[18]。试剂标准曲线(曲线A)采用95%乙腈水溶液逐级稀释配制成0、2、10、50、100、200、500 ng/mL 的系列标准溶液。基质标准曲线(曲线B)用猪尿样品,按照“1.3”方法进行提取净化,用氮吹复溶后的溶液配制相同质量浓度的系列标准溶液。根据基质效应(ME,%)=斜率曲线B/斜率曲线A×100%计算。结果显示,环丙氨嗪和三聚氰胺的ME 分别为86.2%、53.8%,表明猪尿样品存在基质抑制效应,且三聚氰胺的基质效应较为严重。因仪器灵敏度较高,尝试将试剂和基质系列标准溶液稀释10 倍和20 倍。结果表明,稀释10 倍和20 倍后,环丙氨嗪的ME 分别为90.8%、96.1%,基质抑制效应较低,对外标法定量结果影响较小。而三聚氰胺稀释后的ME 仍低于80%,因此三聚氰胺采用同位素内标法定量,以补偿因基质效应引起的响应变化。因此本实验采用将样品溶液稀释10倍进样,以试剂标准曲线定量。
2.6 方法学验证
2.6.1 线性关系与定量下限配制0.2~50.0 ng/mL 的系列标准溶液,环丙氨嗪以峰面积为纵坐标,质量浓度为横坐标绘制标准曲线;三聚氰胺以待测物与内标的峰面积比值为纵坐标,待测物的质量浓度为横坐标绘制标准曲线。结果表明,环丙氨嗪和三聚氰胺均在0.2~50.0 ng/mL 范围内线性关系良好,相关系数(r2)大于0.99。采用样品加标的方式确定定量下限,样品基质中加标1.0µg/kg,采用本方法进行测定,以满足10倍信噪比(S/N)的要求得到方法定量下限为1.0µg/kg。
2.6.2 回收率与相对标准偏差取环丙氨嗪和三聚氰胺空白猪尿样品进行加标回收和精密度实验,加标水平为1.0、2.0、150.0 µg/kg,每个水平重复测定6 次(见表1)。结果显示,环丙氨嗪和三聚氰胺的回收率为78.0%~105%,相对标准偏差(RSD)为1.7%~11%,符合GB/T 27404-2008[19]的要求。

表1 环丙氨嗪和三聚氰胺的回收率及相对标准偏差(n=6)Table 1 Recoveries and RSDs of cyromazine and melamine(n=6)
2.7 实际样品检测
采用本方法对实际猪尿样品进行检测,将UHPLC-Quadrupole-Orbitrap MS 检测的结果与采用LC-MS/MS 测定的结果进行比对。2 个阳性猪尿样品的检测结果如表2 所示,经T检验验证发现,样品中环丙氨嗪的P值分别为0.7 和0.1,三聚氰胺的P值分别为0.1 和0.4,表明UHPLCQuadrupole-Orbitrap MS 和LC-MS/MS 的检测结果不存在显著性差异(P>0.05)。因此,两种检测方法的结果具有可比性。

表2 猪尿样品中环丙氨嗪和三聚氰胺的测定结果Table 2 Determination results of cyromazine and melamine in pig urine samples
3 结 论
本文建立了1%三氯乙酸超声提取,MCX 固相萃取柱净化,超高效液相色谱-四极杆/静电场轨道阱质谱PRM 模式检测猪尿中环丙氨嗪及其代谢物三聚氰胺的方法。该方法灵敏度高,定量结果与三重四极杆质谱结果具有可比性,定性准确,可应用于猪尿样品中环丙氨嗪及其代谢物残留的分析。
