基于织物相吸附萃取技术测定藿香正气水中浸出物及风险评估
2022-01-09杨雨希康天惠孙雪纯杜振霞
杨雨希,康天惠,孙雪纯,杜振霞*
(1.北京化工大学 化学学院,北京 100029;2.首都医科大学 基础医学院,北京 100069)
塑料包装材料因使用方便、成本低等优点被广泛应用于药物的生产、储存及使用环节,但其含有的添加剂及其降解产物在与药品的接触过程中,可能会从接触材料中迁移出来,作为杂质进入药品中,或与药品活性成分形成加合物[1],或改变蛋白质稳定性[2],对药物的质量造成影响,甚至产生毒性,从而影响药品的安全性和有效性[3]。
在药品包装材料的安全性评估中需进行实际药品的浸出试验,然而药品基质通常很复杂,且包装材料迁移的浸出物通常为痕量水平,因此需要选择一种合适的样品前处理技术。目前,浸出物的常用前处理方法主要有液液萃取法(LLE)[4-5]、固相萃取法(SPE)[6]、搅拌棒吸附萃取法(SBSE)[7]等。织物相吸附萃取(Fabric phase sorptive extraction,FPSE)是由Kabir和Furton在2014年提出的一种新型样品前处理技术[8-9]。该技术通过溶胶-凝胶法将吸附剂化学键合在具有大量活性基团的织物基材表面,如棉布、聚酯和玻璃布等天然或人工合成的纤维材料上,形成均匀的超薄涂层。FPSE克服了传统吸附萃取技术的缺点,增大了接触面积,进而提高了富集倍数和富集速度,是一种高效、简单、绿色的微萃取技术,已应用于环境[10]、食品[11]和生物样品[12]的前处理。
藿香正气水是一种常见的中成药,一般灌装在聚乙烯材质的塑料瓶中上市。藿香正气水是采用乙醇和水作为溶剂制成,成品中含有40%~50%的酒精成分[13],较高浓度的乙醇会加速包装瓶中的添加剂及其降解产物向药液迁移,经口服后可能会对人体健康造成潜在危害。因此,有必要开发一种藿香正气水中浸出物的检测方法。
本研究在织物表面键合了聚二甲基硅氧烷(PDMS)涂层用于富集藿香正气水中包装材料的浸出物,探究了提取和解吸条件对回收率的影响。选择乙醇/水(1∶1,体积比)、异丙醇作为可提取物提取溶液,利用超高效液相色谱-四极杆飞行时间质谱(UPLC-QTOF MS)筛查2种提取液中的可提取物,然后利用超高效液相色谱-串联质谱(UPLC-MS/MS)靶向检测实际药液中上述可浸出物的含量,并对可浸出物进行了风险评估。
1 实验部分
1.1 仪器与试剂
Waters Acquity 超高效液相色谱仪、Waters Quattro Premier 三重四极杆质谱仪、Waters Xevo G2-S四极杆飞行时间质谱仪(美国Waters 公司);Milli-Q Advantage A10 超纯水系统(美国Millipore 公司);MTN-2800D 氮吹仪(天津奥特赛恩斯仪器有限公司);SHA-BA 水浴恒温振荡器(华普达仪器有限公司);WH-861 涡旋混合器(太仓市华利达实验设备有限公司)。
色谱纯和质谱纯甲醇(美国Fisher公司);甲酸(色谱纯,美国Aladdin 公司);Milli-QⅠ级超纯水;二氯甲烷、丙酮、盐酸(分析纯,北京化工厂);三氟乙酸(99%)、甲基三甲氧基硅烷(98%);氢氧化钠固体(96%)均购于阿拉丁试剂有限公司;羟基封端的聚二甲基硅氧烷(PDMS-OH,粘度为700~800 cst,Gelest Inc.);无水乙醇(分析纯,天津市大茂化学试剂厂);异丙醇(质谱纯,美国Fisher 公司)。标准品:抗氧剂168、抗氧剂1010、抗氧剂1076、邻苯二甲酸二乙基己酯(DEHP)、芥酸酰胺、棕榈酸、紫外吸收剂UV-1164、紫外吸收剂UV-1577均购于阿拉丁试剂有限公司,纯度均大于95%;用于织物相吸附萃取的织物基材(100%纤维素)购自北京某商场;藿香正气水口服液瓶(规格10 mL,高密度聚乙烯材质)收集于中国的一家制药公司。
1.2 可提取物实验
本研究通过可提取物的全面筛查找出潜在的浸出物。由于藿香正气水含有40%~50%的酒精,因此采用乙醇/水(1∶1,体积比)模拟该药品的溶剂配方进行可提取物实验,乙醇/水(1∶1)也是美国药典新通则USP<665>[14]推荐的提取溶剂。此外,为获得更全面的可提取物清单,选择异丙醇作为提取溶剂以模拟最恶劣的储存环境。
根据ASTM F1980-16《医疗器械无菌材料系统加速实验标准指南》[15],60 ℃是保持聚合物材料完整性的最高温度。根据USP<665>通则,建议将包材在40 ℃下浸泡21 d。根据阿仑尼乌斯公式进行温度和时间的换算[15],相当于在60 ℃下浸泡5.25 d。因此,为模拟更恶劣的提取环境并加快实验进度,本研究采用60 ℃浸泡塑料瓶,在第5.25 d 采样后直接氮气吹干,复溶后过滤膜,采用UPLCQTOF MS进行非靶向筛查。
1.3 织物相萃取介质的制备
1.3.1 纤维布的预处理将一块10.0 cm×5.0 cm 的纯棉纤维布用去离子水清洗数次后,依次浸泡于1 mol/L NaOH溶液和0.1 mol/L HCl溶液中活化1 h,用去离子水洗涤至中性后干燥过夜。
1.3.2 溶胶-凝胶涂层的制备将10 g PDMS、10 mL溶胶-凝胶前驱体甲基三甲氧基硅烷、20 mL二氯甲烷-丙酮(50∶50,体积比)和4 mL 三氟乙酸(含5%水)混合均匀制备成溶胶。将处理过的纤维布浸入溶胶中4 h 后取出,置于烘箱内50 ℃老化12 h,依次用二氯甲烷和甲醇漂洗后干燥1 h,裁剪成2.0 cm×2.0 cm的小片待用。
1.4 织物相萃取步骤
萃取前将涂层织物置于10 mL 甲醇-乙腈(50∶50,体积比)混合物中,超声清洗10 min,以清洁并活化涂层。将5 mL 藿香正气水样品、织物萃取相和聚四氟乙烯磁力搅拌子置于玻璃样品瓶中,以700 r/min 萃取30 min 后,取出织物并轻轻地压在纸巾上使其干燥。将织物用1 mL 乙腈超声洗脱10 min,洗脱液氮吹至干,用1 mL 乙腈复溶,过0.22µm 有机滤膜后上机检测。将涂层织物再次按上述步骤清洁并干燥后,保存于密闭容器中待用。
1.5 色谱-质谱条件
1.5.1 色谱条件采用Waters ACQUITYTMUPLC CORTECS C18(100 mm × 2.1 mm,1.6 µm)色谱柱;流动相:由A 相(甲醇)和B 相(正模式为含0.1%甲酸水溶液,负模式为水)组成;梯度洗脱程序:0~3 min,60%~99% A;3~9 min,99% A;9~9.1 min,99%~60% A;9.1~11 min,60% A;流速:0.3 mL/min;柱温:30 ℃;进样量:2µL。
1.5.2 飞行时间质谱条件电离模式:ESI+或ESI-;检测模式:MSE;扫描范围:m/z50~1 200;毛细管电压:3 kV(+)、2.5 kV(-);离子源温度:120 ℃;脱溶剂气温度:450 ℃;脱溶剂气流速:800 L/h;锥孔气流速:50 L/h;锥孔电压:30 V;碰撞能量:20~60 eV;实时校正LockSpray:亮氨酸脑啡肽作为实时校正溶液,正模式m/z556.277 6、负模式m/z554.262 0,每30 s 切换1 次进行质量数的校正。
1.5.3 三重四极杆质谱条件电离模式:ESI + 或ESI-;扫描方式:选择离子模式(SIR);扫描范围:m/z50~1 200;毛细管电压:3 kV(+)、2.5 kV(-);离子源温度:120 ℃;脱溶剂气温度:350 ℃;脱溶剂气流速:600 L/h;锥孔气流速:50 L/h;锥孔电压:30 V。
2 结果与讨论
2.1 可提取物的确证
乙醇/水(1∶1)和异丙醇的可提取物样品与空白样品的基峰强度(BPI)色谱图分别如图1 和图2 所示。采用自建谱库结合互联网在线检索的方法对可提取物进行鉴定。本实验室的自建谱库由300 种聚合物添加剂的详细信息组成,包括化学结构、理论质量、保留时间和碎片离子等。UNIFI 软件可通过在线图书馆自动对可提取物进行识别,并与谱库中的物质相匹配。对于自建聚合物谱库中无法直接识别的化合物,利用UNIFI 软件通过联网筛查的方式进行鉴定。在2 种浸泡液中共检出33 种化合物,结果如表1所示。
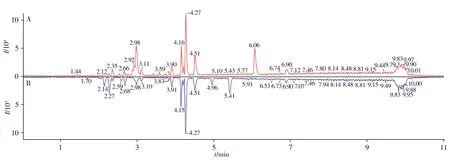
图1 乙醇/水(1∶1)的可提取物样品(A)与空白样品(B)的UPLC-QTOF MS基峰强度(BPI)色谱图Fig.1 UPLC-QTOF MS base peak intensity(BPI)chromatograms of extractables in ethanol/water(1∶1)(A)and blank sample(B)
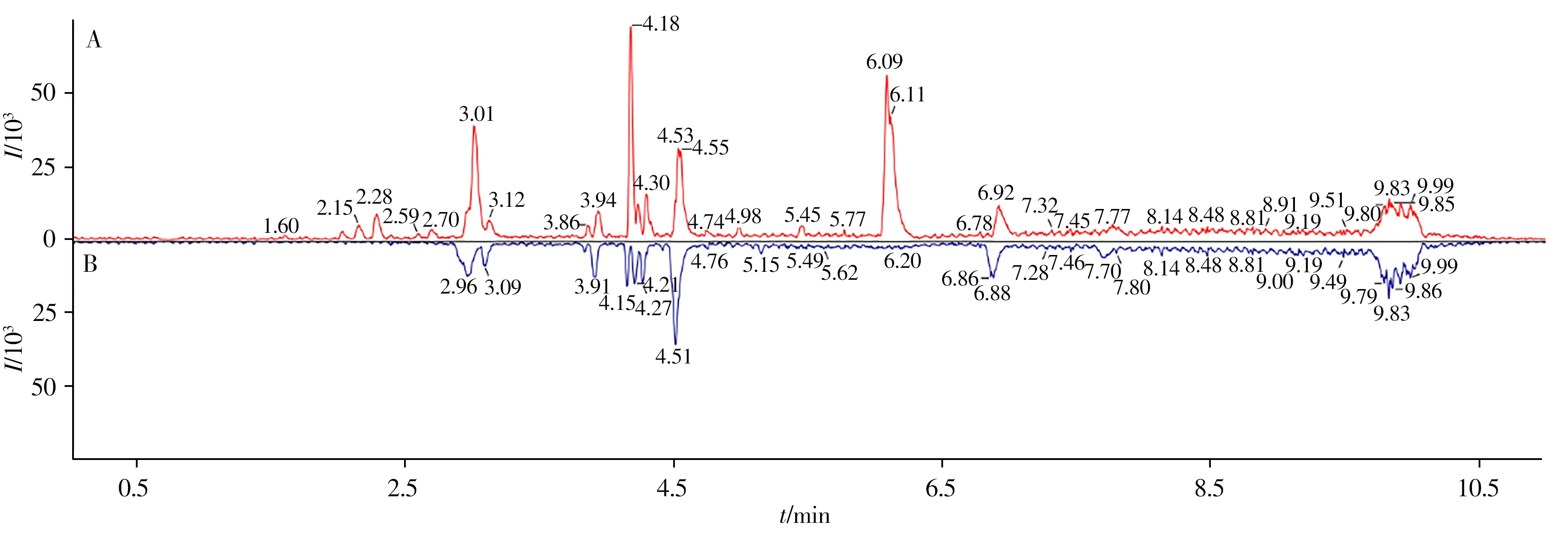
图2 异丙醇可提取物样品(A)与空白样品(B)的UPLC-QTOF MS基峰强度(BPI)色谱图Fig.2 UPLC-QTOF MS base peak intensity(BPI)chromatograms of extractables in isopropanol(A)and blank sample(B)
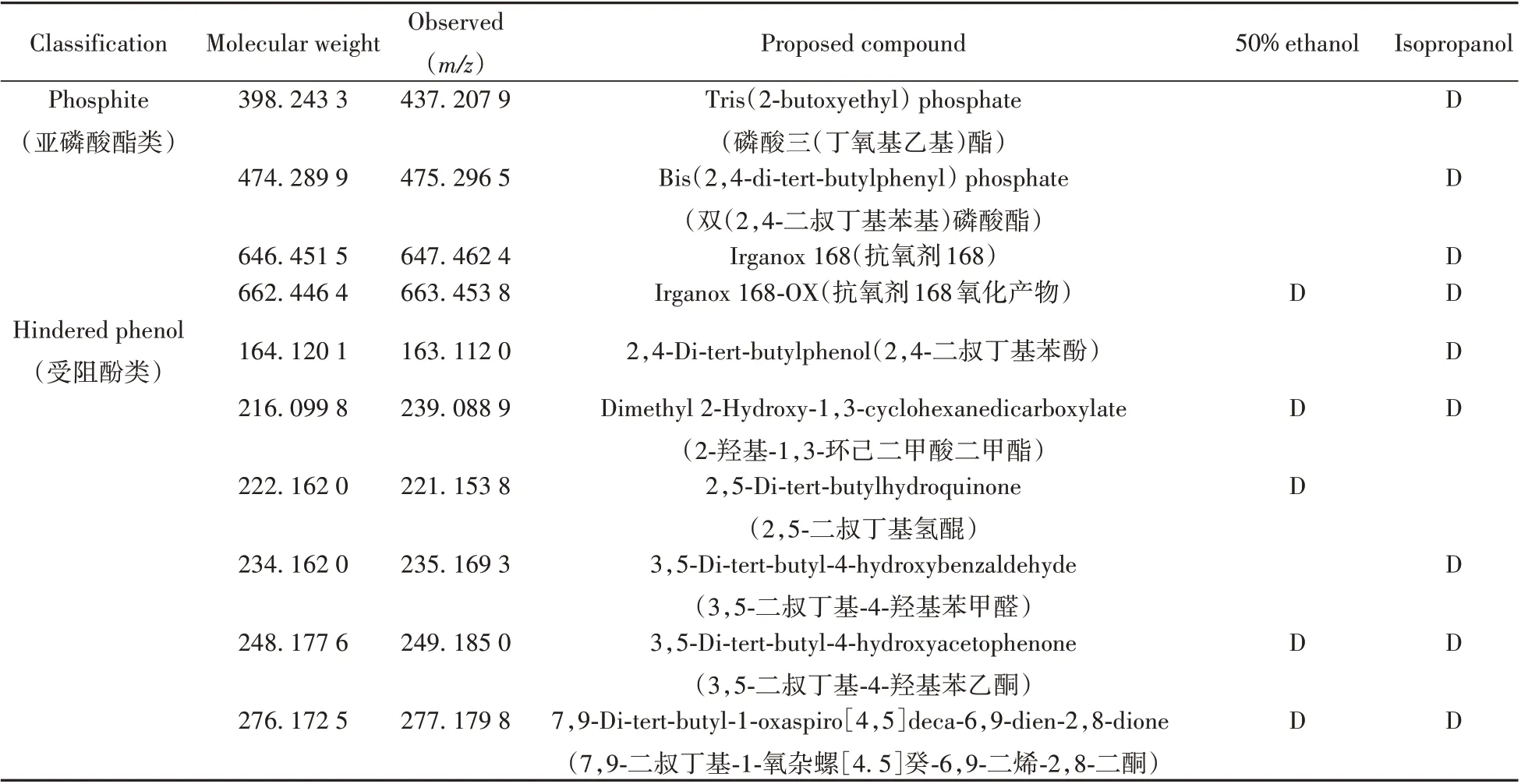
表1 可提取物筛查结果Table 1 The possible identification results of the extractables

(续表1)
2.2 溶胶-凝胶涂层的表征
溶胶-凝胶吸附涂层类似于海绵状的多孔三维网络,具有高孔隙率和良好的渗透性,通过分子间相互作用(伦敦色散力、偶极-偶极相互作用和氢键作用等)达到吸附的目的。经PDMS 修饰后,纯棉布的实物图如图3A 所示,在涂覆后的4 cm2棉布上PDMS 的平均负载量为19.46 mg,相对标准偏差(n=6)为4.0%。如图3B、C、D 所示,扫描电镜(SEM)图显示了在溶胶-凝胶吸附剂涂覆前后织物基材表面形态的变化。由SEM 图可见,涂覆前棉纤维表面光滑,涂覆后表面变得粗糙,说明PDMS 键合在织物表面,展现了较高的吸附容量。

图3 PDMS涂覆后的纯棉布实物图(A),未涂覆的织物基材放大1 000倍(B)、溶胶-凝胶PDMS涂层放大1 000倍(C)、溶胶-凝胶PDMS涂层放大3 000倍(D)的扫描电镜图Fig.3 Image of sol-gel coated FPSE media(A),and scanning electron microscopy of uncoated cellulose fabric substrate at 1 000×magnification(B),sol-gel PDMS coated FPSE membrane at 1 000×magnification(C)and 3 000×magnification(D)
2.3 萃取与解吸参数的优化
将可提取实验中鉴别的33 种可提取物列为潜在的浸出物清单,通过UPLC-MS/MS 靶向检测藿香正气水中33种潜在的浸出物。由于可提取物中大多数为添加剂的降解产物,无法得到标准品,因此将其按结构分成亚磷酸酯类、受阻酚类、邻苯二甲酸酯类、脂肪酸类、酰胺类、三嗪类及其他。在每类中选择1~2 种化合物或与其结构类似的物质作为内标物进行方法学优化,所选择的8 种标准品为抗氧剂168、抗氧剂1010、抗氧剂1076、邻苯二甲酸二乙基己酯、芥酸酰胺、棕榈酸、UV-1164、UV-1577。优化实验在加标量为100 ng/mL的药液中进行,每个萃取参数重复3次实验取平均值。采用加标回收率评估提取效率。
2.3.1 提取时间考察了提取时间(5~60 min)对提取效率的影响,结果显示,随着提取时间的延长,8种分析物的回收率逐渐增加,在30 min时达到最大。但进一步延长提取时间(30~60 min)会导致某些化合物的回收率降低,可能是由于少量的分析物又回溶到样品溶液中。因此,选择最优提取时间为30 min。
2.3.2 离子强度通常,向样品中添加氯化钠提高样品的离子强度,有利于疏水性的目标化合物向萃取介质迁移(盐析效应),从而提高萃取效率。但较高的盐含量会增加样品的粘度,使传质速率降低,从而减慢目标物的提取速率[16]。因此,实验考察了氯化钠含量(0%、2%、5%、10%)对萃取效率的影响。结果表明,随着氯化钠含量的增加,8 种分析物的回收率均呈下降趋势,说明盐的加入不利于萃取。因此,在后续的实验中不加盐。
2.3.3 解吸溶剂种类比较了3 种溶剂对解吸效率的影响,使用甲醇(MEOH,相对极性0.762)、乙腈(ACN,相对极性0.46)或甲醇-乙腈混合物(50∶50)对目标分析物进行反萃取。如图4 所示,乙腈显示出最好的反萃取能力,因此实验选择乙腈为解吸溶剂。
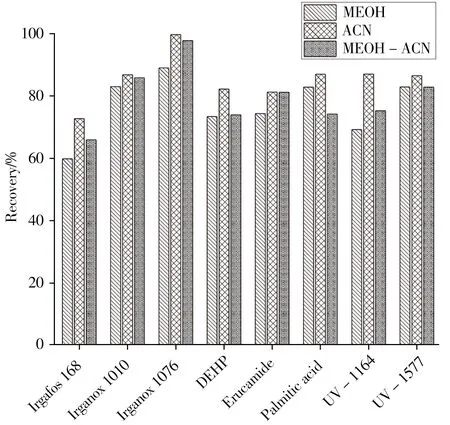
图4 解吸溶剂种类对分析物回收率的影响Fig.4 Effect of desorption solvent type on recoveries of the analytes
2.3.4 解吸溶剂体积为了获得更高的洗脱效率,考察了解吸溶剂乙腈体积为0.5、1、2 mL(小于0.5 mL乙腈不足以浸没萃取介质)时对解吸效率的影响。由图5可知,乙腈体积为0.5 mL时,8种目标物的回收率较低。乙腈体积为1 mL时可将目标化合物解吸完全,若继续增加解吸溶剂体积,回收率未明显增加。因此,实验选择解吸溶剂乙腈的体积为1 mL。
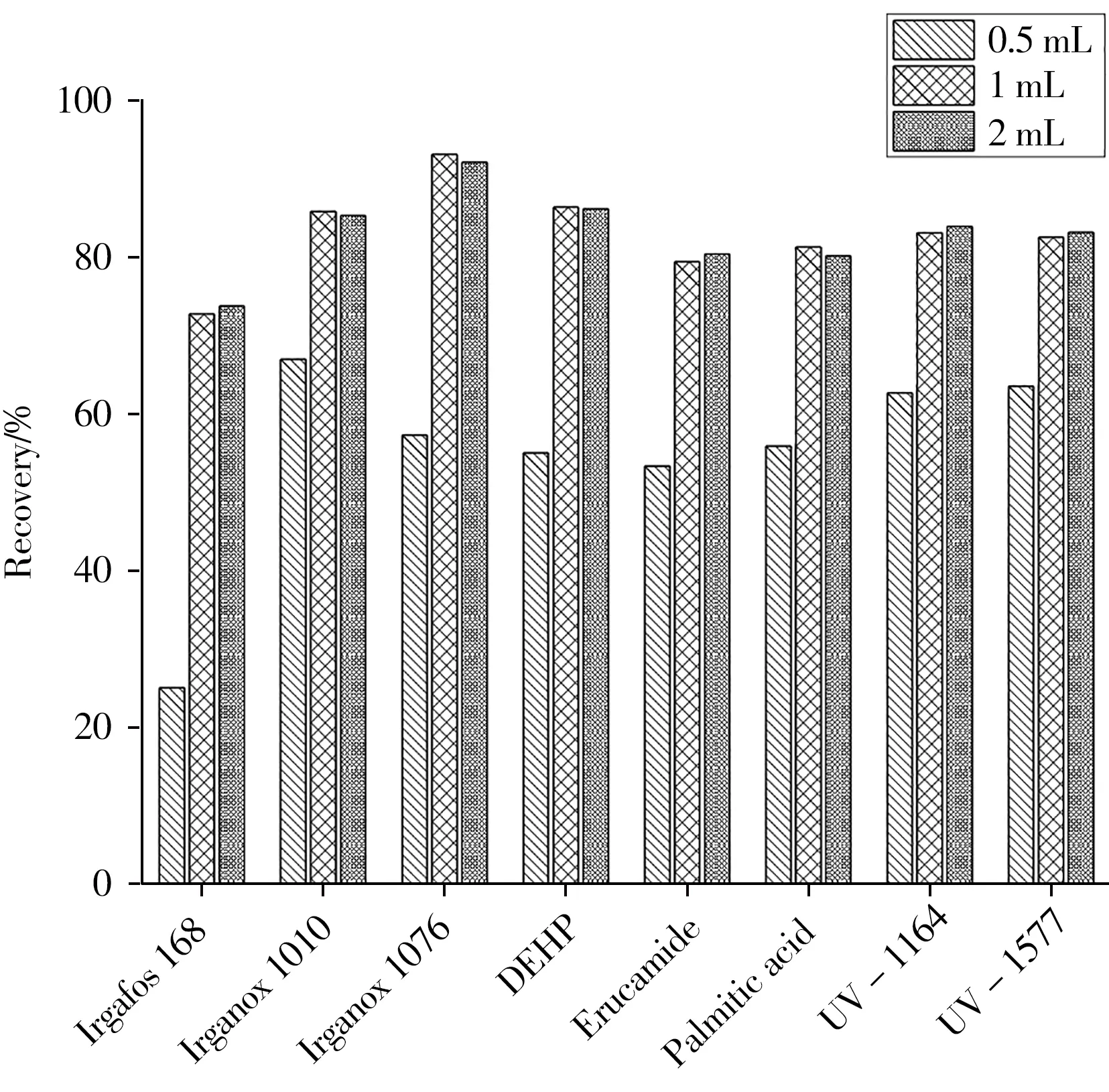
图5 解吸溶剂体积对分析物回收率的影响Fig.5 Effect of desorption solvent volume on recoveries of the analytes
2.3.5 解吸时间考察了解吸时间(5、10、15、20 min)对洗脱效率的影响。结果显示,当解吸时间从5 min增至10 min时,目标分析物的回收率显著增加;若进一步延长解吸时间,回收率则保持恒定或略有降低,可能是因为分析物在FPSE介质上发生了再吸收。因此,选择最佳解吸时间为10 min。
2.4 线性关系、检出限与定量下限
为消除基质的影响,建立基质校正标准曲线进行定量分析。取5 mL 空白样品,采用“1.4”方法进行前处理得到空白样品基质,向其中添加8 种标准品得到系列质量浓度的基质标准溶液,采用UPLC-MS/MS 检测。8 种标准品(1 mg/L)的色谱图如图6 所示。以质量浓度为横坐标(x,µg/L),峰面积为纵坐标(y),绘制8 种标准品的标准曲线。结果显示,8 种标准品的线性关系良好,相关系数r≥0.996 1,检出限(LOD,S/N≥3)为0.5~1.5µg/L,定量下限(LOQ,S/N≥10)为1.5~4.0µg/L(表2)。

表2 8种标准品的线性范围、线性方程、相关系数、检出限(LOD)和定量下限(LOQ)Table 2 Linear ranges,linear equations,correlation coefficients,limits of detection(LOD)and limits of quantitation(LOQ)of the 8 standards
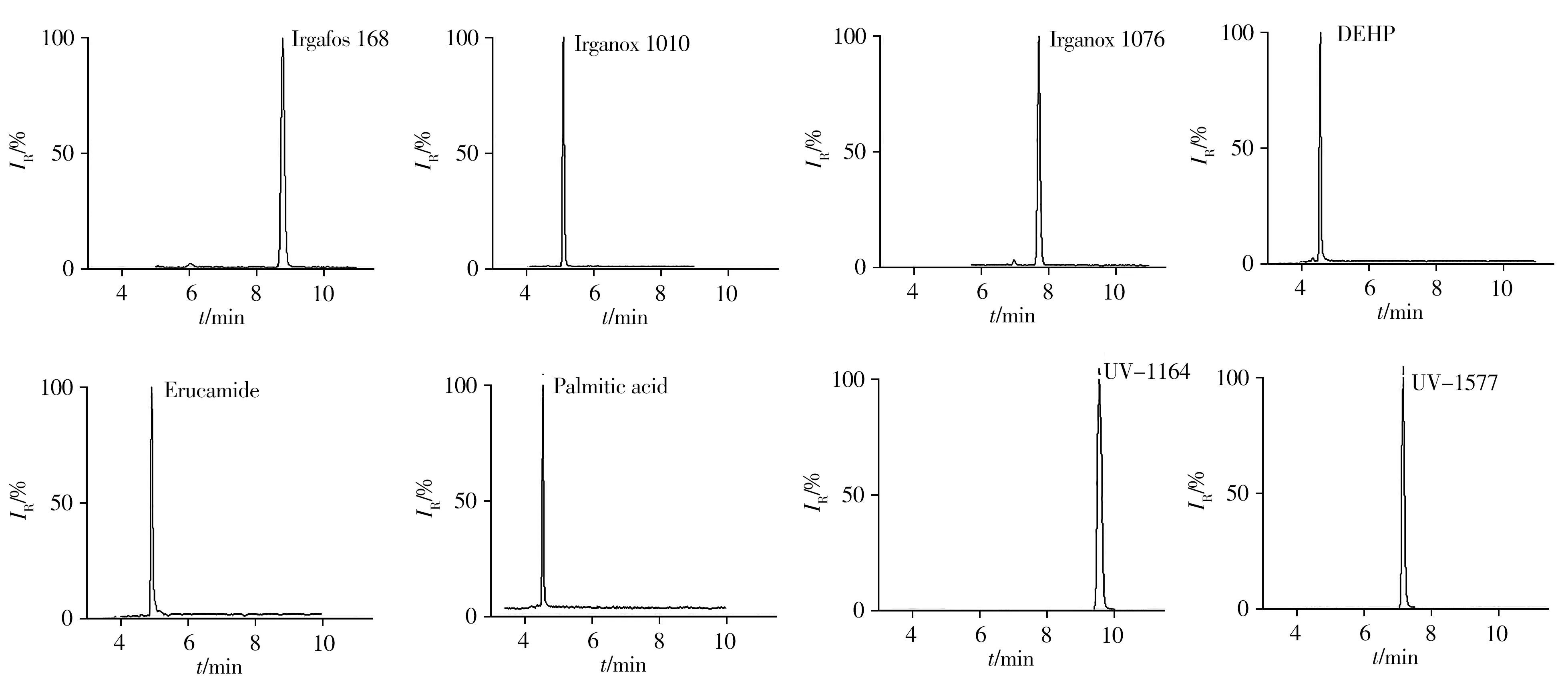
图6 8种标准品(1 mg/L)的色谱图Fig.6 Chromatograms of the 8 standards(1 mg/L)
2.5 回收率与相对标准偏差
为验证方法的准确性,进行了加标回收实验。向空白药液中添加10、50、100µg/L 3个水平的8种标准品,采用优化后的FPSE方法对加标后药液进行萃取,每个加标水平制备3组平行样。由表3可见,8种标准品的平均加标回收率为73.8%~98.2%,RSD为0.98%~8.4%。
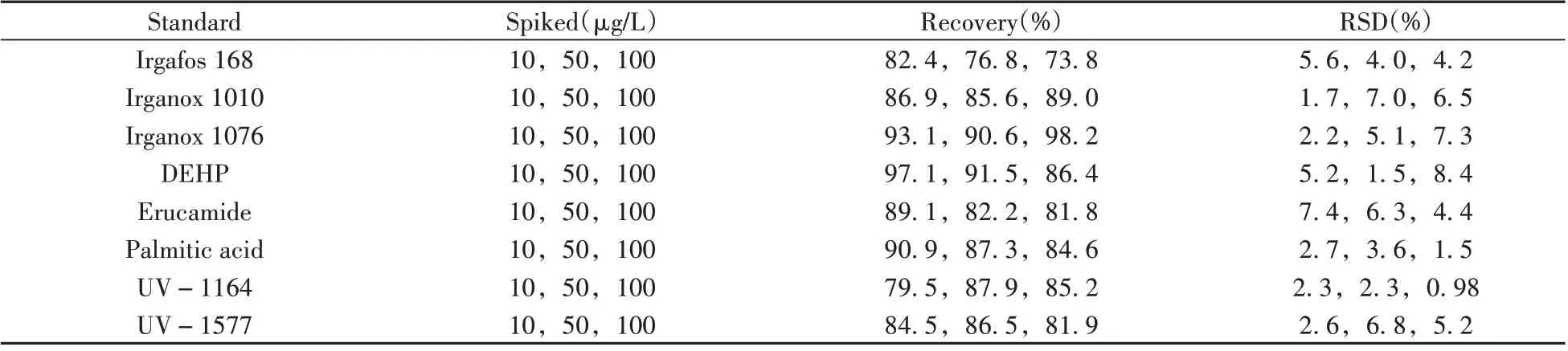
表3 8种标准品的平均加标回收率和相对标准偏差(n=3)Table 3 Average spiked recoveries and relative standard deviations(RSD)for the 8 standards at 3 spiked levels(n=3)
2.6 实际样品的检测
取5 mL 塑料包装瓶内的藿香正气水置于玻璃样品瓶中,经“1.4”方法前处理后,通过超高效液相色谱-三重四极杆质谱靶向测定在“2.1”可提取物实验中筛查出的物质。药液中浸出物的含量均根据“2.4”中所选标准品的标准曲线计算得到,定量结果见表4。结果共检出4 种浸出物,其中抗氧剂168(Irgafos 168)的氧化产物Irgafos 168-OX的质量浓度最高,其余3种物质的质量浓度较低。
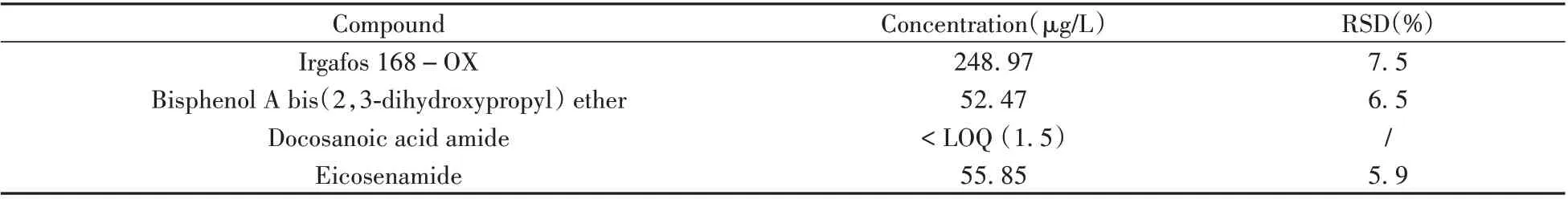
表4 藿香正气水中浸出物的定量结果Table 4 Quantitative results of leachables in Huoxiang Zhengqi liquid
2.7 风险评估
本实验基于毒理学评价软件Toxtree 和T.E.S.T.对4 种浸出物进行了风险评估。首先通过Toxtree 软件得到致癌性预测(遗传毒性和非遗传毒性),确定4种浸出物均无遗传毒性。然后通过T.E.S.T.软件得到浸出物的大鼠LD50。根据公式(1),可由LD50预测得到无明显作用剂量(NOEL)[17],根据ICH Q3C 指导原则,NOEL可用于推导成人每日允许暴露量(PDE),见公式(2)[18]。
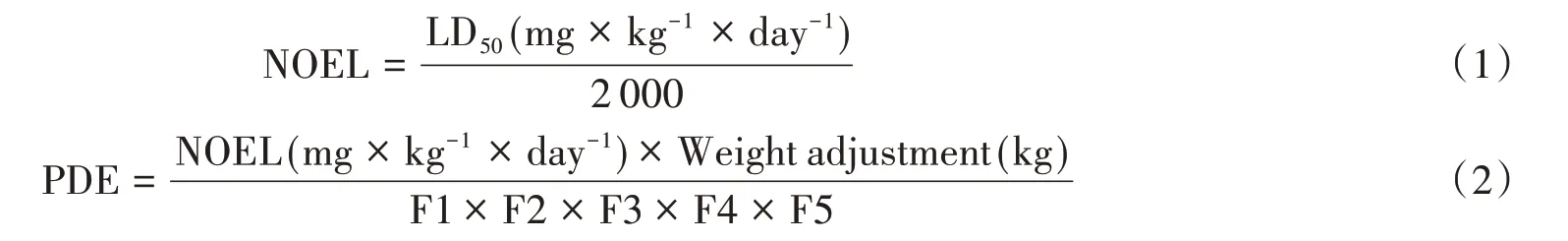
式中,Weight adjustment取50 kg,F1~F5均为不确定因子,根据ICH Q3C指导原则中不确定因子的取值规则[18],在本研究中的取值依次为5、10、10、10、1。根据药方说明,每人每次服用藿香正气水10 mL,一日两次,依据“2.6”实际药液中浸出物的近似含量计算患者的每日实际摄入量(见表5)。结果表明4 种浸出物的每日摄入量均未超过PDE 值,因此藿香正气水中的浸出物对人体而言是相对安全的。
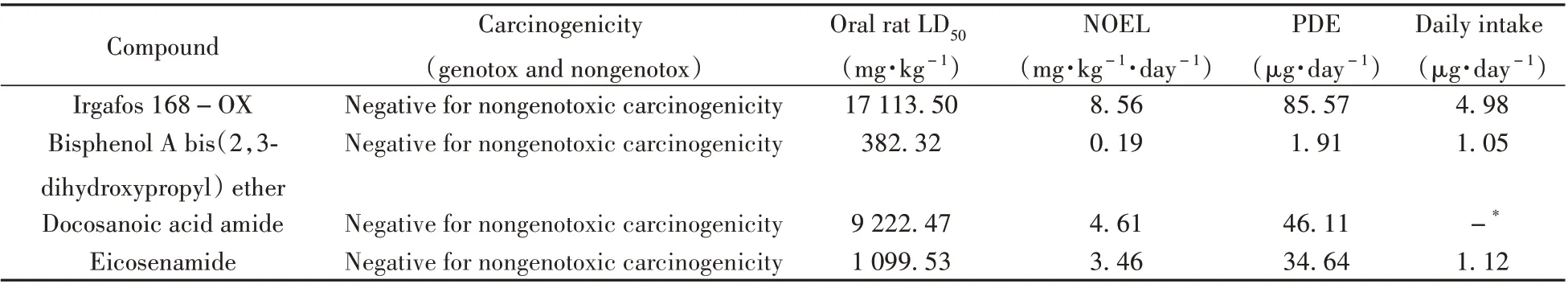
表5 藿香正气水中4种浸出物的毒理学评价结果Table 5 Toxicological assessment results of 4 kinds of leachables in Huoxiang Zhengqi liquid
3 结 论
本研究建立了塑料包装瓶藿香正气水中可提取物与浸出物的快速筛查、定量分析方法。首先通过可提取物实验得到了包装瓶的浸出物清单,然后通过织物相萃取技术结合超高效液相色谱-串联质谱联用对8 种标准品进行了方法学验证,最后对浸出物进行了检测,共检出4 种化合物。风险评估实验发现4 种浸出物含量处于相对安全的范围。本文建立的方法具有操作简单、灵敏度高、重现性好的优点,也可用于其他药品中浸出物的检测,为药品生产企业选择合适的药品包装材料提供了参考依据。
