一个Leber先天性黑蒙症家系的分子诊断与产前诊断
2021-12-15余秀蓉刘伊楚兰风华王志红
余秀蓉,刘伊楚,兰风华,曾 健,林 娟,王志红
0 引 言
Leber先天性黑蒙症(Leber congenital amaurosis, LCA)是1869年由德国眼科医师Theodor Leber首先报道[1]。该病是最严重的遗传性视网膜疾病,可导致婴儿在出生后1个月内完全丧失视力[2]。患儿常表现为眼球震颤、固视障碍、畏光、按压眼球、黑蒙性瞳孔等眼部症状。视网膜电图(electroretinogram, ERG)表现为a、b波平坦,甚至消失,具有诊断价值。LCA具有遗传和临床异质性,多数为常染色体隐性遗传,也有少数报道为常染色体显性遗传,基因突变谱在不同的种群间存在差异[3]。LCA常见的致病基因主要有CEP290、GUCY2D、CRB1、RPE65、IMPDH1、AIPL1和RPGRIP1等,约50%~60%的LCA患者可以通过基因检测明确病因[4]。本研究采用二代测序方法对一个LCA家系的先证者进行基因诊断,并通过PCR扩增及直接测序方法对该家系的高危胎儿进行产前分子诊断,并回顾了相关文献。
1 资料与方法
1.1 研究对象先证者,女,2014年12月生,1岁,因“生后不能视物”就诊于我院眼科,眼科检查:角膜透明,瞳孔直径2.5 mm,对光反应存在,眼球震颤,眼球内陷,指眼征阳性。标准全视野ERG:双眼未引出正常波形,双眼各振幅熄灭。眼底照相提示视网膜色素变性,显示周边视网膜色素沉着,见图1。患儿肾脏彩超、生化全套、动脉血气分析、尿常规、尿微量蛋白检查均正常,患儿听力正常,智力、运动发育正常。根据患者病史及检查结果,临床拟诊LCA。先证者父母既往身体健康,无LCA家族史。先证者母亲再次妊娠16周行遗传咨询,并要求对先证者行LCA相关的基因诊断以及对所怀胎儿进行产前诊断。家系图见图2。本研究经本院伦理委员会批准(批准号:2016019),先证者父母均签署知情同意书。
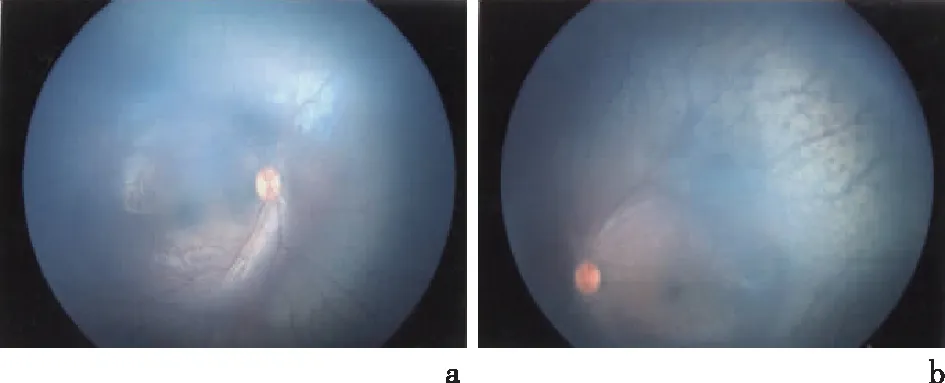
a:右眼;b:左眼图示周边视网膜色素沉着图1 Leber先天性黑蒙症患者眼底照相
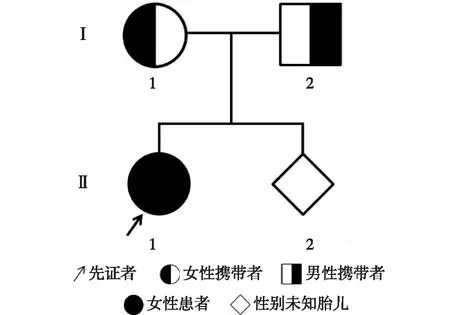
Ⅰ-1:先证者母亲;Ⅰ-2先证者父亲;Ⅱ-1:先证者;Ⅱ-2:胎儿图2 Leber先天性黑蒙症家系图
1.2 方法
1.2.1 基因组DNA提取采集先证者及其父母外周血各2 mL,先证者母亲妊娠20周,由产科医师穿刺抽取其羊水约10 mL。使用DNA提取试剂盒(Qiagen公司)分别提取该家系的外周血和羊水细胞的基因组DNA,并将DNA样本置-20 ℃冰箱保存备用。
1.2.2 二代测序先证者外周血送深圳华大临床检验中心进行LCA相关的基因Panel(AIPL1、CABP4、CEP290、CRB1、CRX、GUCY2D、IQCB1、LCA5、LRAT、RD3、RDH12、RPE65、RPGRIP1、SPATA7、TULP1、IMPDH1、OTX2、KCNJ13和NMNAT1)检测。主要包括DNA文库制备,芯片捕获和富集,测序仪完成基因检测,基因突变分析并结合致病变异和正常人基因组等数据库筛选出LCA相关的变异。
1.2.3 Sanger测序针对先证者在IQCB1第11外显子发现的突变,按照参考文献[5],由铂尚生物技术(上海)合成覆盖突变位点的引物,上游序列:5′-AGATTGCACAACAGCAGCAG-3′,下游序列:5′-TCATCACGTAGCTAGAAAAGTTGG-3′。扩增产物片段长度为389 bp,包含IQCB1基因c.1090C>T突变位点。对先证者及其父母和高危胎儿的基因组DNA进行PCR扩增,配置PCR反应体系,设置循环参数:95 ℃/5 min;95 ℃/30 s,56 ℃/30 s,72 ℃/30 s,共35个循环;72 ℃/5 min。PCR产物送铂尚生物技术(上海)有限公司进行Sanger测序。
1.2.4 母体基因组污染排除取胎儿羊水细胞和父母外周血的基因组DNA,按照参考文献[6]用Identifiler试剂盒(ABI公司)进行PCR扩增,使用3100 Avant遗传分析仪(ABI公司)检测15个短串联重复序列(short tandem repeat, STR),再用GeneMapper Software v3.7软件鉴定STR位点。根据母亲独有的STR位点是否在羊水细胞样本中出现,判断是否存在母体基因组污染[7]。
2 结 果
2.1 二代测序LCA相关基因Panel测序发现先证者IQCB1基因c.1090C>T(p.R364X)纯合突变。该突变为已知致病性突变。
2.2 Sanger测序测序结果证实先证者存在IQCB1基因c.1090C>T纯合突变,先证者父母和高危胎儿的IQCB1外显子11基因测序显示均为c.1090C>T杂合突变,见图3。
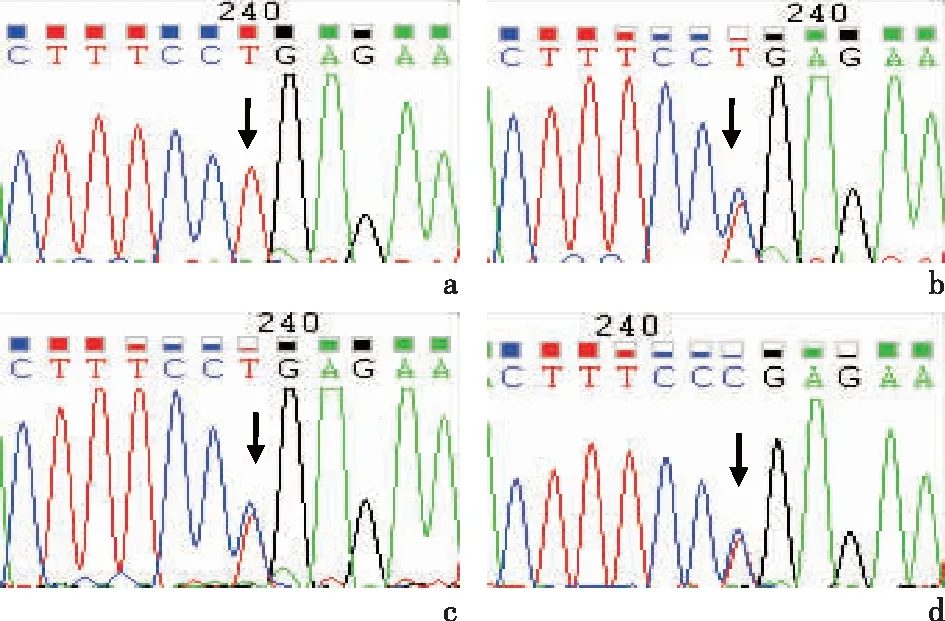
a:先证者(Ⅱ-1)检出IQCB1基因c.1090C>T纯合突变;b:先证者父亲(Ⅰ-2)检出IQCB1基因c.1090C>T杂合突变;c:先证者母亲(Ⅰ-1)检出IQCB1基因c.1090C>T杂合突变;d:胎儿(Ⅱ-2)检出IQCB1基因c.1090C>T杂合突变图3 Leber先天性黑蒙症家系IQCB1基因外显子11部分序列图
2.3 母体基因组DNA污染的排除胎儿及其父母基因组DNA的STR位点分析结果显示:母亲基因组DNA独有的STR位点均未在胎儿羊水细胞中出现,羊水细胞基因组DNA来自独立个体,因此,可以排除母体基因组DNA污染。
3 讨 论
IQCB1(IQ motif containing B1)基因,也称NPHP5(nephrocystin-5)基因,是眼-肾综合征(Senior-Loken syndrome, SLSN)最常见的致病基因,该病呈常染色体隐性遗传[8]。近年来IQCB1基因突变报道主要见于LCA和SLSN患者,截至2019年4月,人类基因突变数据库(The Human Gene Mutation Database)已收录44种突变类型,包括19种无义与错义突变、6种剪接突变、12种小片段缺失突变和7种小片段插入突变。通过二代测序,本研究发现先证者存在IQCB1基因c.1090C>T(p.R364X)致病性纯合突变。该突变由美国Khanna等[9]于2009年首次报道,Tong等[5]和Yu等[10]在3个中国SLSN患者检测出该纯合突变。通过一代测序验证,先证者的父母亲都携带有IQCB1基因c.1090C>T杂合突变,他们没有眼部及肾脏的病变,符合常染色体隐性遗传的特征。
IQCB1基因位于3q13.3,有15个外显子,编码由598个氨基酸残基组成的蛋白,其中包含一个螺旋结构域(340-373氨基酸残基)和两个钙调蛋白IQ结合域(294-323和387-416氨基酸残基)。该蛋白主要表达在连接感光细胞的纤毛和肾小管上皮细胞的初级纤毛,负责上皮细胞的完整性。有研究表明该蛋白合成障碍会导致视网膜变性和肾囊肿形成[8]。c.1090C>T(p.R364X)为无义突变,该突变提前引入终止密码子,使得mRNA转录提前终止,产生由363个氨基酸组成的异常截短蛋白。由于该截短蛋白丧失了原有蛋白完整的螺旋结构域和钙调蛋白IQ结合域,导致蛋白功能缺陷,从而致病。
IQCB1基因突变所致的SLSN典型临床表现是视网膜和肾脏的病变,而无其他器官损害。该型患者100%会出现视网膜病变,重者早期出现LCA,轻者表现为轻度视力损害和视网膜色素变性,部分患者可仅表现为早发视网膜色素变性或LCA而无肾功能衰竭[11-12]。2016年,美国犹他大学的Ronquillo等[13]对IQCB1基因敲除小鼠进行实验研究,发现IQCB1基因编码的蛋白对小鼠光感受器外节层形成至关重要,但对小鼠肾脏和小鼠胚胎成纤维细胞纤毛的形成不是必须的。有研究表明,IQCB1基因突变患者发生肾功能衰竭的年龄差异较大,早者3岁发病,迟者可延迟到50岁,因此,仅表现为LCA的患者发生肾功能衰竭的风险高,若未能早期诊断,有可能突然死于水电解质失衡[14]。目前IQCB1基因已成为临床表型为LCA和(或)肾单位肾痨(nephronophthisis, NPHP)患者的共同筛查基因,可明显提高SLSN的临床诊断率。
本研究中先证者目前仅表现出LCA而无肾脏病变,需要定期进行尿常规、尿微量蛋白、肾功能等监测以达到早发现早治疗、预防肾衰竭的目的。目前,虽然有报道基因治疗方法应用该病的临床治疗[15],但是产前诊断/胚胎植入前产前诊断仍然是预防此类患儿出生的主要途径。本研究中胎儿检测结果为IQCB1基因c.1090C>T杂合突变,根据遗传规律,该胎儿出生后患SLSN的风险低。先证者母亲于妊娠36周2天顺产1健康女孩,LCA /NPHP相关的眼部及肾脏检查均未发现异常。
