孟德尔随机化方法在肾脏疾病研究中的应用
2021-09-18逯静茹综述刘志红审校
逯静茹 综述 刘志红 审校
随着基因组学等各种“组学”技术的发展以及高通量测序技术等生物技术的进步,生物医学数据的积累呈指数级增长,开创了数据驱动医学研究的新时代。如何从海量生物学数据中挖掘出最有用的信息,是对生命科学以及医学研究的一个巨大挑战。大数据和人工智能算法的融合使人类从前所未有的视角来了解危险因素与疾病风险间的因果关联,为系统解读疾病的病因及发生发展机制提供新思路。
孟德尔随机化方法(Mendelian randomization,MR)是将人类复杂性状和疾病风险相关的基因组数据整合到传统的流行病学研究设计中,借助个体的遗传信息来推断危险因素(暴露)与疾病风险(结局)的因果关联[1-2],从而反映两者之间的真实联系。而且,MR弥补了传统流行病学研究的不足,尤其在随机对照试验难以实施、观察性研究由于混杂因素或反向因果关联而使结果偏倚时具有重要应用价值[2]。
以基因组为代表的多组学大数据仍将不断涌现,亟需加速向医学应用领域渗透,才能进一步服务于医疗决策和疾病的防诊治管理。基于基因组数据推断相关危险因素与疾病风险间的因果关联,有效促进了大数据向临床应用的转化。目前,MR已广泛应用于医学研究。本文综述了MR的设计、分析步骤、研究类型、局限性及其在慢性肾脏病(CKD)、糖尿病肾病(DN)方面的应用,为更深层面认识疾病以及科学防治开拓思路。
MR研究
研究设计随着人类基因组计划、人类基因组单体型图计划(HapMap)、千人基因组计划的顺利完成,研究人员已获得人类基因组中常见变异位点的详细图谱。在此基础上,全基因组关联研究(GWAS)得以迅猛发展,复杂疾病或性状相关的基因组数据激增[3],催生了MR的出现及应用。
MR设计的核心是工具变量的使用。工具变量的概念起源于计量经济学,后来广泛被医学研究所采用[4]。工具变量是指与目标危险因素相关,但与其他因素或混杂因素无关的可测量的变量。在MR研究设计中,将遵循孟德尔遗传定律的遗传变异作为工具变量,指代无法测量的待研究危险因素。通过分析遗传变异与危险因素、遗传变异与疾病风险的关联,进而推断危险因素与疾病风险之间的因果关联[5]。Katan[6]于1986年首次提出这一概念。由于研究基于孟德尔第二遗传定律,即在人类配子形成过程中,亲代等位基因随机分配给子代,所以称之为孟德尔随机化研究。
MR的分析模型为“遗传变异-危险因素-疾病风险”。已知遗传变异与危险因素强相关,如果该危险因素与疾病风险有因果关联,那么携带这些遗传变异也会改变疾病风险。MR使用相反的思路进行推断:已知遗传变异与危险因素强相关,通过分析遗传变异是否与疾病风险有关,从而推导危险因素与疾病风险的因果关系。
传统的医学统计学及流行病学研究存在一定局限性。传统的相关性分析包括相关分析和回归分析两种统计学手段。其基于既定参数模型进行统计分析所得到的关联性结果,仅能说明变量间存在伴随协变的趋势,无法直接推论变量间的因果关联[7]。另外,由于常受到潜在混杂因素及反向因果关联的影响,传统观察性研究的结果也无法直接反映因果关系[8]。随机对照试验(RCTs)是检验医学相关暴露与结局之间因果关系的金标准。然而,RCTs实施难度较大,常受到医学伦理的限制,且有较高的失败率[9]。
MR可以弥补上述传统方法的不足。MR依赖于减数分裂过程中遗传变异的自然、随机分配,使得遗传变异在人群中随机分布[10]。个体在出生时自然、随机地携带了影响危险因素的遗传变异,如导致低密度脂蛋白胆固醇(LDL-C)水平升高的遗传变异或不遗传该变异。在给定人群中,根据是否携带该遗传变异将人群进行分组,进而比较两组间结局的发生情况。这种基于个体与生俱来的遗传变异进行分组的方式是完全随机的,不受人群其他特征、环境、社会经济地位等混杂因素的干扰,且因果时序合理,类似于随机对照试验中的随机分组过程。两个携带不同等位基因亚组间的疾病风险差异将表明该风险因素(如LDL-C)对疾病的因果效应。
在临床应用中,MR不仅有助于了解疾病病因,还能为疾病治疗提供新策略。例如,几项大型的观察性研究均表明血浆LDL-C水平降低与冠心病发生率降低相关,但这些研究难以避免LDL-C相关因素的混杂作用。研究表明前蛋白转化酶枯草杆菌蛋白酶Kexin 9型(PCSK9)基因的变异与LDL-C水平的降低有关。PCSK9是一种糖蛋白,主要在肝脏中合成,可与肝脏表面的LDL受体结合,减弱肝脏代谢血浆LDL-C的能力,从而导致LDL-C水平升高。MR使用PCSK9基因变异分析LDL-C与冠心病的关系。证据表明遗传变异与LDL-C水平相关,同时也与冠心病风险相关。该研究为LDL-C对冠心病的因果关联提供支持证据,而且提示PCSK9可能是降低LDL-C的新靶标[11]。目前,已证实PCSK9单克隆抗体作为新型降脂药可将LDL-C降至前所未有的水平,并且对心血管疾病具有保护作用。
研究步骤MR设计主要包括三个步骤:(1)确定待研究的危险因素(暴露)和疾病风险(结局),可在观察性研究设计中检测两者的相关性。(2)选择合适的遗传变异[由GWAS获得与危险因素显著相关的单核苷酸多态性(SNP)]作为工具变量。(3)MR统计学分析,检测与危险因素相关的遗传变异是否也与疾病风险相关,以评估危险因素对疾病风险的因果效应。
上述工具变量(遗传变异)的选择是研究的关键步骤。MR研究的工具变量(遗传变异)必须满足三个核心假设:①工具变量(遗传变异)必须与危险因素密切相关;②工具变量(遗传变异)不得与影响“危险因素-疾病风险”关系的混杂因素相关;③工具变量(遗传变异)只能通过危险因素与疾病风险相关联,而不能通过其他途径影响疾病风险(图1)[5]。当任一假设不成立时,将难以准确推断因果关联。弱工具变量、遗传变异的多效性、人群分层、连锁不平衡等因素均可能导致假设的不成立,因此在进行MR统计学分析前需对三个核心假设进行评估以保证结果的可靠性[1]。
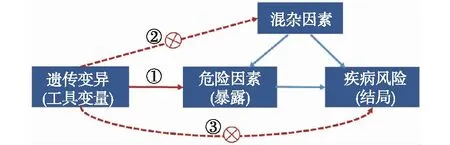
图1 孟德尔随机化方法中工具变量的三个核心假设[5]
研究类型目前常用的MR研究类型包括单样本、两样本、双向和两阶段MR等[1-2]。经典研究设计是单样本MR,指研究人群来自同一研究样本。可以使用个体水平数据,在同一样本中测量危险因素(暴露)、疾病风险(结局)和遗传变异(工具变量)。两样本MR指遗传变异和危险因素之间以及遗传变异和疾病风险之间的关联数据来自相同人群的两个独立样本。通常使用既往GWAS研究的汇总数据,也可使用个体水平的数据。两样本MR要求两个样本具有相似的年龄、性别和种族分布。该方法无需评估危险因素和疾病风险的关联信息就可以进行因果推断[1]。双向MR主要用于危险因素和疾病风险的因果方向不确定时,从两个方向分别进行了MR分析,以确定两者因果关联的方向。两阶段MR可以检测危险因素和疾病风险之间的因果关系被感兴趣的中间变量介导的程度。两阶段MR方法的发展源于表观遗传流行病学,用于研究甲基化介导暴露与医学相关结局之间因果关联的程度[12]。目前也常使用其他变量如生物标志物等作为中间变量进行评估。
局限性虽然MR优于传统的观察性流行病学研究,但其也存在一定的局限性:(1)难以获取合适的遗传变异。尽管已发现了大量与复杂性状相关的遗传位点,但仍有一些感兴趣的危险因素由于缺少GWAS研究或公开数据资源等原因而难以获得相关的遗传变异。即使获取了遗传变异,也可能由于不满足3个核心假设而无法有效推断因果关联。(2)统计能力不足。统计能力不足会减低结果的准确性。MR研究中统计能力的决定因素包括遗传变异的频率、遗传变异对危险因素的效应量以及研究样本量。将多个变异整合为多基因风险评分或增加样本量有助于提高统计能力。(3)结果不易解读。大多数遗传变异的生物学机制尚不清楚,这使得难以解释因果关联中潜在的作用机制。结合生物学知识、生物信息学分析和后续的基础实验将有助于研究结果的解读。(4)Beavis效应:复杂疾病或性状通常与大量基因位点相关,而GWAS研究仅报道最显著的小部分遗传变异,这可能高估了遗传变异与危险因素的关联,从而影响MR研究中的因果推断。
MR在CKD的应用进展
全球CKD发病率高达10%,其可能导致终末期肾病(ESRD),并与预期寿命缩短有关[13]。CKD的治疗选择有限,主要集中在控制血压、减少蛋白尿和并发症。大量MR研究显示,多种生物标志物、伴发疾病以及生活方式与CKD间存在因果关联,为CKD患者的科学管理提供新的理论依据。
生物标志物与CKD 尿酸、血脂等多个生化指标的异常与CKD的因果关系尚不明确,借助MR方法使用生物标志物相关的遗传变异作为工具变量探讨其与CKD的因果关联具有重要意义,相应研究已取得了一定进展。既往观察性研究一致表明血尿酸水平与CKD风险之间有很强的正相关性[14],但两者之间是否存在因果关系仍然未知。Jordan等[15]使用多个欧洲队列进行了大样本MR分析(n>400 000),从GWAS荟萃分析(n=110 347)中选取26个与尿酸相关的SNP作为工具变量,推断其与CKD的因果关联。MR分析未发现尿酸水平与CKD风险有因果关联的证据(所有P>0.05),表明单纯降低尿酸水平可能并不会降低CKD发生风险。这一结论否定了既往观察性研究得到的关联结果,主要归因于MR在很大程度上避免了潜在混杂因素对结果的干扰。
既往流行病学研究显示血脂水平与CKD风险的相关性仍存在诸多争议,并且难以证明因果关联。为探究血脂水平与CKD的因果关联,Lanktree等[16]使用了全球血脂遗传合作组的188 577例欧洲个体的GWAS数据和CKD遗传学合作组的133 814例欧洲个体的GWAS数据进行了两样本MR研究,发现高密度脂蛋白胆固醇(HDL-C)水平每升高17 mg/dl导致估算的肾小球滤过率(eGFR)升高0.8%(95%CI 0.4%~1.3%)、CKD风险降低15%(OR=0.85; 95%CI 0.77~0.93)。这表明较高的HDL-C水平可能与肾功能改善及CKD风险降低有因果关联[16-17]。
CKD存在明显的性别差异,男性的肾功能下降速度快于女性,并且在CKD透析前的各个阶段,男性的死亡率更高。然而,尚不清楚CKD性别差异的原因。为此,Zhao等[18-20]借助英国生物银行队列(UK Biobank)的GWAS数据进行了一系列的MR研究,分别选择与睾酮[18]、性激素结合球蛋白(SHBG)[19]、胰岛素抵抗[20]强相关的SNP作为工具变量,探讨其在179 916例男性以及212 079例女性中与CKD的性别特异性关联。结果表明睾酮、SHBG、胰岛素抵抗可能是男性CKD的潜在病因,而与女性无关。作者借助MR方法证实了多个导致CKD性别差异的原因,为CKD病因学提供了新线索。
其他疾病与CKD MR除了探讨CKD与其生物标志物的病因联系,也探讨了与其他疾病的因果关联。传统观察性研究表明甲状腺功能减退与CKD风险增加有关,而肾脏疾病也可能引起甲状腺功能减退[21-22]。然而,甲状腺功能减退与CKD之间的因果关系及方向尚无定论。Ellervik等[23]使用女性基因组健康研究和CKD遗传学合作组的基因组数据进行双向MR研究以推断因果方向。遗传证据表明甲状腺功能减退使甲状腺刺激素(TSH)和甲状腺过氧化物酶抗体(TPOAb)增加,从而导致eGFR减少和CKD风险增加。反之,则因果关联不成立。这为CKD的发生发展机制及防治管理提供了重要参考。
生活行为习惯与CKD 生活习惯的改变可能会对公众健康产生重大影响,因此探索各种生活行为习惯与疾病的因果关联具有重大意义[24-25]。在全球范围内,每天消费超过20亿杯咖啡。流行病学研究显示经常喝咖啡的人CKD发生风险较低[26],但是这些研究可能存在高血压、肥胖、吸烟等混杂因素的干扰。Kennedy等[24]基于UK Biobank和CKD遗传学合作组的GWAS数据,选择与咖啡消费相关的25个独立SNP作为工具变量,评估饮用咖啡与CKD的因果关联。MR研究结果表明每天多喝一杯咖啡,CKD 3~5期的风险降低16%(OR=0.84; 95%CI 0.72~0.98),并且与更高的eGFR水平相关(β=0.022;P=1.6×10-6)。该研究提供了咖啡对肾功能有益的依据。
MR在DN的应用进展
DN是糖尿病常见的并发症,其发病机制较为复杂,现有的治疗干预措施只能部分延缓DN的发生和发展,约有50%的DN患者可进展至ESRD。借助MR研究进行因果推断可帮助揭示DN的发病机制,并为新的治疗策略提供参考依据。
肥胖被认为是DN的独立危险因素,但尚不明确两者之间是否存在因果关系。为此,Todd等[27]从欧洲最大的GWAS研究[28]中选取32个与体质量指数(BMI)相关的SNP位点,针对上述位点对6 049例1型糖尿病患者进行基因分型,进一步计算加权遗传风险评分并将其作为BMI的工具变量,从而进行MR分析以推断BMI与DN的因果关联。遗传证据显示BMI每升高1 kg/m2会导致ESKD风险增加43%(OR=1.43, 95%CI 1.20~1.72)、DN风险增加33%(OR=1.33, 95%CI 1.17~1.51),表明肥胖与DN具有因果关联。
DN筛查主要基于反映肾小球损害的白蛋白排泄率(AER)或eGFR。然而,肾小管损伤也可能在DN进展中起重要作用。研究证实,急性肾损伤分子1(KIM-1)是急性肾损伤或CKD的生物标志物[29],但尚未在1型糖尿病患者中研究KIM-1与肾功能的潜在因果关系。Panduru等[30]基于1 573例1型糖尿病患者的GWAS数据进行了MR研究,选择与尿液KIM-1水平强相关的rs2036402作为工具变量,推断其与肾功能的因果关联。遗传证据表明KIM-1水平升高可导致eGFR降低,且独立于糖尿病病程和AER(β=25.044;P=0.040),提示KIM-1与1型糖尿病患者的肾功能间存在因果关联。
其他MR研究的遗传学证据表明补体C3[31]、循环同型半胱氨酸[32]水平升高与DN风险增加有因果关联,而且尿触珠蛋白[33]水平升高可促进DN进展。然而,可溶性晚期糖基化终产物受体[34]、血清尿酸浓度[35]与DN进展无直接因果关联。值得注意的是,目前DN相关的MR研究主要集中在1型糖尿病中[27,30,34-35],而2型糖尿病患者的相应研究较少[32-33],主要由于受到了GWAS及相应基因组数据的限制。期待在2型糖尿病患者中开展更多相关研究,为其病因解读及防治策略提供新见解。
小结:MR是从遗传学依据中进行因果推断,借助个体的遗传标志来反映相关危险因素和疾病风险间可能的因果联系。通常由GWAS获得与危险因素强相关的遗传变异作为工具变量,进而推断其与疾病风险的因果关联。该方法弥补了传统研究的不足,在很大程度上减少了混杂因素对结果的影响,避免了反向因果关系和回归稀释偏倚。随着后GWAS的展开,MR在医学研究领域具有广阔的应用前景。
