基于组基础模型的HIV /AIDS发病和死亡纵向资料双轨迹分析*
2021-07-07高洪艳
郭 剑 高洪艳 王 媛
【提 要】 目的 介绍组基础模型双轨迹分析原理,以HIV/AIDS发病和死亡纵向资料演示。方法 建立HIV/AIDS发病、死亡组基础模型,确定最优轨迹组数目和发展轨迹形态,将发病、死亡组基础模型参数带入双轨迹分析,以概率形式反映1990-2015年195个国家或地区HIV/AIDS发病、死亡轨迹组间关联性。结果 组基础模型将HIV/AIDS发病和死亡都分为4组:高水平、中高水平、中低水平、低水平。双轨迹分析表明,发病高水平组成为死亡高水平组概率为66.7%,成为死亡中高水平组概率为33.3%;发病中高水平组成为死亡中高水平组概率为80%,成为死亡高水平组概率为20%。发病低水平组、中低水平组与死亡相应组别100%对应。结论 组基础模型双轨迹分析能以概率形式反映两测量结局发展轨迹间关联程度和离散程度。
组基础模型双轨迹分析(group-based model dual trajectory analysis,GBMDTA)被用来分析两个测量结局纵向资料的关联性。在纵向研究中,研究者可能关心两个不同测量指标的关联程度。同时期的,如成年人血压与血脂;不同时期的,如儿童期肥胖和成年血压。传统用于表述两个不同但存在关联的测量指标间关系的统计量主要是相关系数、比值比等。这样最多只关注两个时期的统计方法,无法充分利用纵向研究数据信息。而且超过两个时期的纵向数据能反映的远不止一条线性发展轨迹,这也是传统的汇总式统计量无法有效反映的。GBMDTA通过联接两个不同指标的发展轨迹,以概率形式评价两者关联程度。最早,由Nagin等人用于分析儿童期过渡活跃与成年期焦虑的关系[1],在心理学领域有所应用。而公共卫生领域罕有报道。本研究旨在介绍GBMDTA原理,并以HIV/AIDS发病和死亡纵向资料演示,以推动其在公共卫生领域的应用。
资料与方法
1.资料来源
资料来源于全球疾病负担研究(global burden of disease,GBD 2017)195个国家或地区1990年、1995年、2000年、2005年、2010年、2015年HIV/AIDS年龄标化发病率和死亡率。
2.基本原理[2]
(1)GBMDTA原理
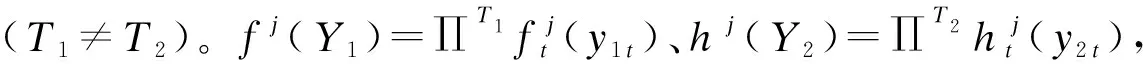
似然函数:假设Y1的J个轨迹组与Y2的K个轨迹组存在概率上的联系,以j和k,Y1与Y2为条件的分布相互独立,则Pjk(Y1,Y2)=fj(Y1)hk(Y2)。因此,Y1与Y2的非条件似然函数为每个个体的Pjk(Y1,Y2)合计,并且每个这种条件分布以πjk(属于Y1的j个轨迹组和Y2的k个轨迹组的成员联合概率)为权重。
(1)
公式(1)中πjk=πk|jπj,因此公式(1)也可写作:
(2)
公式(2)中的似然函数由两个部分按顺序组成,Y1的每个j组与Y2的每个k组通过条件概率πk|j联接。公式(2)自然反映了Y1在时间上先于Y2的顺序。然而,无论孰先孰后,公式(2)也有另一种对等形式,通过条件概率πj|k联接每个k组与每个j组。对于每个个体的似然函数公式可表达为:
(3)
描述两个结局轨迹间关联的概率用πj|k、πk|j和πjk表示,说明两结局发展轨迹的重叠程度。公式(1)~(3)中涉及πj、πk、πj|k,πk|j和πjk,计算过程介绍如下:
①πj表示一个随机选取的个体Y1属于某一亚组j的概率,即总体中第j个亚总体的比例:
(4)
其中,θj,j=1,2,…,j,是由不带协变量的多项式logit模型估计所得的参数。因各组πj相加等于1,只需计算J-1个θj。
②πk表示一个随机选取的个体Y2属于某一亚组k的概率。
(5)
πjk=πk|jπj
(6)
公式(5)表示Y2的k个轨迹组中每一个组的成员概率。公式(6)说明公式(5)中每一部分是属于轨迹组k和j的成员联合概率,πjk=πk|jπj,即在给定j轨迹组条件下,属于第k组的概率。为计算πk需要对Y1的j个组的联合概率求合计。因为个体属于Y2的某一轨迹组k的同时,也属于Y1的J个轨迹组中的一组,J个轨迹组的联合概率πjk的合计等于πk。
③πj|k可用于反映Y1的j轨迹组对Y2特定的轨迹组k的贡献。即在给定某些k轨迹组时,个体属于特定j轨迹组的概率。
(7)
④πk|j在给定轨迹组j条件下,个体属于轨迹组k的概率。
(8)
其中,γk|j,j=1,2,…,J,k=1,2,…,K,是由不带协变量的多项式logit模型估计所得的参数。
公式(8)需要计算J×K个概率。对每一个Y1的J个轨迹组,都有K个转换概率,对应Y2的K个轨迹组之一。因此,每个亚组j需要估计K-1个参数,每一个对应Y2的K-1个轨迹组之一。第K组的转换概率能够通过1减其他K-1个组的概率得到。因此,公式8需要估计J×(K-1)个参数。即为Y1的J个轨迹组中的每个亚组估计K-1个参数。
GBMDTA的两个概念模型。在限制模型中Y1的每个轨迹都与Y2的轨迹单独关联。研究者可以假设特定Y1与Y2轨迹一一对应。在全模型中Y1与Y2间的关联限制被去除,取而代之的是采用概率描述轨迹间的关联。这种多方面关联允许模型反映轨迹间联接特征的模式,研究者也不必假定两个不同结局的关联形式(图1)。本研究采用全模型的形式演示GBMDTA分析过程。
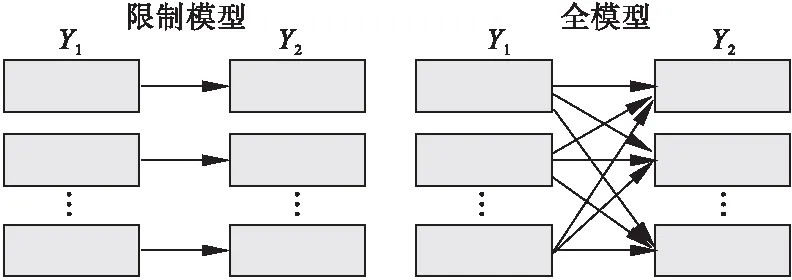
图1 概念模型
(2)分析步骤
建立单指标组基础模型,确定最优轨迹组数目和发展轨迹形态,依据BIC(接近0者拟合优度最好)筛选不同模型[3]。确定模型后,将两个单指标组基础模型所得参数代入GBMDTA。
(3)统计软件
采用SAS 9.4统计软件包的PROC TRAJ过程实施GBMDTA拟合。
结 果
1.单指标组基础模型拟合
HIV/AIDS年龄别标化发病率和死亡率为正偏态分布资料。因此,本研究将其进行标准化正态变换后再分析。
模型选择过程:从1组开始逐步增加轨迹组数目,尝试各轨迹组形态包括:常数、线性、2次曲线、3次曲线。将BIC作为筛选标准选择最佳模型(BIC越接近0越好),具体如下:
HIV/AIDS发病率:1组 BIC=-1664.22(N=1170),BIC=-1662.42(N=195);2组 BIC=-1023.95(N=1170),BIC=-1016.78(N=195);3组 BIC=-897.68(N=1170),BIC=-888.72(N=195);4组BIC=-892.64(N=1170),BIC=-880.10(N=195)。因分4组时BIC最接近于0,最终确定发病率分为4个轨迹组,从低到高各组轨迹形态分别为:线性、线性、线性、3次曲线。地理分布:高水平组(4.6%)多位于非洲南部;中高水平组(2.7%)多位于非洲东部;中低水平组(6%)多位于非洲中西部;低水平组(86.6%)位于世界各地(图2)。发病率GBM估计结果见表1。

表1 HIV/AIDS发病率GBM参数估计结果
HIV/AIDS死亡率:1组BIC=-1664.22(N=1170),BIC=-1662.42(N=195);2组BIC=-1093.07(N=1170),BIC=-1086.80(N=195);3组BIC=-940.96(N=1170),BIC=-932.00(N=195);4组BIC=-884.44(N=1170),BIC=-872.79(N=195)。因分4组时BIC最接近于0,最终确定死亡率分为4个轨迹组,从低到高各组轨迹形态分别为:线性、线性、线性、2次曲线。地理分布范围与发病率相近:高水平组(3.6%)、中高水平组(4.1%)、中低水平组(7.7%)、低水平组(84.7%)(图2),死亡率GBM估计结果见表2。
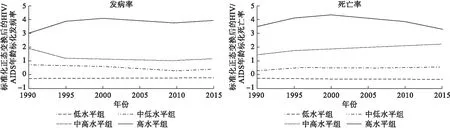
图2 1990-2015年全球HIV/AIDS发病率、死亡率轨迹分组

表2 HIV/AIDS死亡率GBM参数估计结果
2.HIV/AIDS发病与死亡GBMDTA
我们将表1、表2中发病率、死亡率GBM估计结果所得参数带入GBMDTA中作为模型参数估计的初始值,拟合模型。
因概率基于发病率轨迹组,表3中每列合计为1。发病率高水平组成为死亡率高水平组可能性为66.7%,成为死亡率中高水平组可能性为33.3%;发病率中高水平组成为死亡率中高水平组概率为80%,成为死亡率高水平组概率20%。发病率低水平组、中低水平组分别与死亡率相应组别100%对应(表3)。
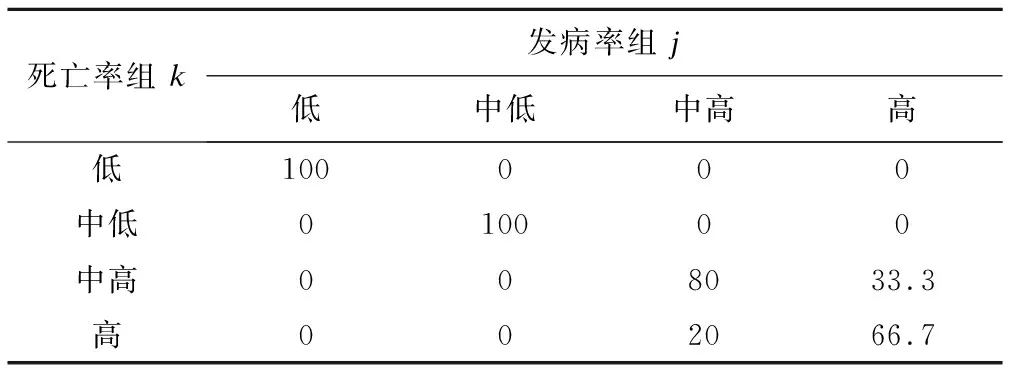
表3 基于发病率组j条件的死亡率组k的概率(πk|j,%)
因概率基于死亡率轨迹组,表4中每行合计为1。死亡率高水平组85.7%归因于发病率高水平组,14.3%归因于发病率中高水平组;死亡率中高水平组57.1%归因于发病率中高水平组,42.9%归因于发病率高水平组。死亡率低水平组、中低水平组分别100%归因于发病率的低水平组、中低水平组(表4)。

表4 基于死亡率组k条件的发病率组j的概率(πj|k,%)

①πk=3,4|j=3为表1中发病率中高组对死亡率中高组、高组概率合计80%+20%=100%。
②πj为总体估计比例,前面发病率组基础模型时4组比例,低水平组πj=1=86.6%,中低水平组πj=2=6%,中高水平组πj=3=2.7%,高水平组πj=4=4.6%。
③πk=3,4可根据公式(5)利用πj和表1中的πk|j求得:
πk=3=πk=3|j=1πj=1+πk=3|j=2πj=2+πk=3|j=3πj=3+πk=3|j=4πj=4=0×86.6%+0×6%+80%×2.7%+33.3%×4.6%=3.7%
同样可求得:
πk=4=3.6%
πk=3,4=πk=3+πk=4=3.7%+3.6%=7.3%。

因此,死亡率的中高水平组和高水平组37%归因于发病率的中高水平组。
表5是发病率轨迹与死亡率轨迹的联合概率,总计4×4=16个联合概率的合计为1。结果表明,84.9%的国家同时属于发病率和死亡率的低水平组,7.9%属于发病率和死亡率中低水平组,3.1%属于发病率和死亡率高水平组,其余以此类推(表5)。
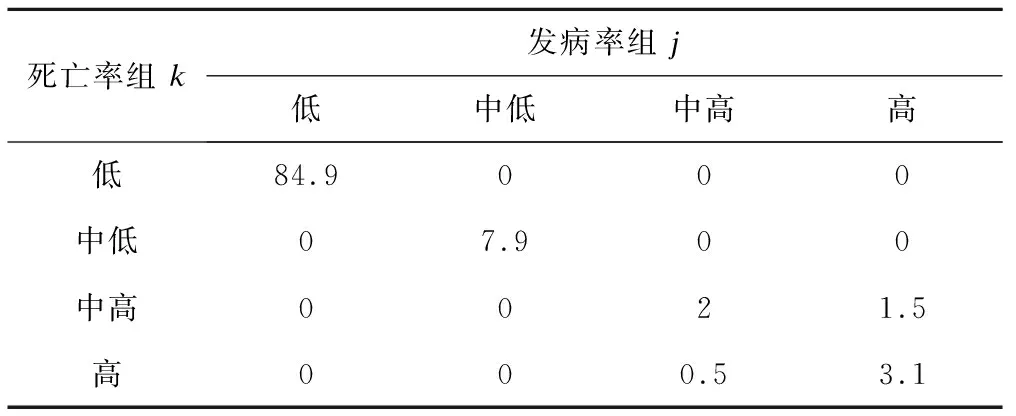
表5 发病率组j和死亡率组k的联合概率(πjk,%)
3.HIV/AIDS发病率和死亡率地理分布
图3为全球195个国家发病率、死亡率地理分布图。HIV/AIDS发病、死亡水平在全球分布并不均衡。以非洲中部、南部最为严重,其余各地较低[4]。从中可见HIV/AIDS发病、死亡分布范围相似,与GBMDTA结果基本一致。

图3 2015年全球195个国家或地区HIV/AIDS发病率和死亡率
讨 论
本研究结果表明,HIV/AIDS发病率、死亡率都被分为4个轨迹组。GBMDTA表明,发病率高水平组成为死亡率高水平组可能性为66.7%,成为死亡率中高水平组可能性为33.3%;发病率中高水平组成为死亡率中高水平组概率为80%,成为死亡率高水平组概率20%。发病率低水平组、中低水平组分别与死亡率相应组别100%对应。大多数国家(84.9%)同时属于发病率和死亡率低水平组。很少国家(3.1%)的国家同时属于发病率和死亡率高水平组。
GBMDTA是建立在单指标组基础模型之上的分析方法,通过事先将测量结局划分为不同等次的轨迹组描述纵向资料的发展轨迹,区分了资料的异质性。传统统计方法是假设所有个体来自同一研究总体,即反映异质性的亚总体的平均相关程度。事实上某些亚总体可能只存在较强相关,其他亚总体可能存在微弱关联。
另一方面,GBMDTA能反映两个结局测量的轨迹组趋势,每个轨迹组中的成员概率,跨轨迹组间的关联成员概率。采用轨迹组的形式总结感兴趣结局的发展轨迹以发掘纵向资料的特征,更专注于两个变量间动态重叠的幅度。相比之下,传统统计方法,最多只关注两个时期,仅反映同期、非同期关联,未能充分利用耗资庞大的纵向研究数据信息。而且,GBMDTA通过总结每种结局的轨迹组的跨组关联,以概率的形式表达,而非单一的总结性统计量,不仅能反映轨迹组间平均趋势,还能阐释对平均趋势的离散程度,为反映两个变量间发展过程的内在联系模式提供了丰富的信息。
GBMDTA通过πj|k,πk|j和πjk三组概率可以描述两个发展轨迹的关联程度,可以帮助研究者探索以下问题:Y2特定轨迹组的成员概率是否独立于Y1的轨迹组成员概率?这有助于评价Y1与Y2发展轨迹的关联,即两变量间平均趋势的关联程度。本研究中各国家的HIV/AIDS发病率与死亡率轨迹就存在着密切的对应关系。另一方面,通过Y1与Y2联合成员概率,判定分布于极端轨迹组的亚群大小。这有助于评价针对特定极端亚组开展干预项目的成本效益。本研究中发病率高水平组与死亡率高水平组联合概率3.1%,主要包含位于非洲南部的7个国家。相比低流行区,这些地区为HIV/AIDS流行的重灾区,开展针对性的防控项目成本效益更为合理。
GBMDTA仅限于两个结局的关系,无法满足分析两个以上结局随时间变化关系的需求。因此,有研究者建议采用结构方程模型思路[5],构建两个以上结局测量发展过程的线性或非线性潜发展模型(latent growth model with multiple growth process),这一思路与GBMDTA一样也包括多测量结局同时存在的平行发展过程(parallel growth processes)和具有前因后果的顺序发展过程(sequential growth process)[6-7]。然而,随着测量结局数量增多,结构方程模型复杂性也升高,要估计的参数也增加,模型所需样本量也增大。
GBMDTA能在区分总体异质性的前提下探索两个结局各轨迹亚组间的关联性。在公共卫生领域中,可用于纵向研究探索两研究因素的关联程度,如一定时期内某地高危性行为频率与HIV/AIDS流行情况关系,或个体腰臀比变化对血压影响等。
