ChemCloser:一个基于路径与片段匹配的药物设计软件
2021-06-21汪伟亮邓卫平
汪伟亮, 邓卫平
(华东理工大学药学院,上海新药设计重点实验室,上海 200237)
新药研发是一个资源密集、时间周期长(10~20 a)、投资高(5~26亿美元)的过程[1-2]。如何提高开发效率、降低开发成本是大家关心的问题。在先导化合物(或已知药物)的基础上,设计与先导化合物有类似的空间结构、带有相同药效基团的“me-too”化合物,这样的化合物可以与先导化合物作用于同一个酶,产生类似的药效。这种方式显然可以降低开发难度,提高新药开发的效率和成功率。如何确切把握先导化合物的空间结构,设计结构新颖、价键合理、空间结构与先导化合物相同或相似的骨架是设计“me-too”化合物的关键问题。
目前发展的药物设计方法多是针对某一个蛋白结合位点来生成可能的药物分子结构,包括:蛋白质结合位点的原子或片段连接法:LUDI (Ludwigshafen)[3];片段生长连接法:LEA3D (Ligand by Evolutionary Algorithm of 3 Dimensions)[4], LigBuilder (Ligand Builder)[5-6], eSynth (electrical Synthesis)[7];按顺序构建法:LEGEND[8],SPROUT[9];随机连接法:CoG(Compound Generator)[10], Flux (Fragment-based Ligand bUilder reaXions)[11];分解已知活性分子后再拼接的方法:eMolFrag (electrical Molecular Fragments)[12],molBLOCKS[13], SPARK[14]等。为解决设计“me-too”化合物的新型骨架的问题,本文开发了一个可以提取结构式的长链,用已知的简单片段对长链进行匹配,最后进行拼接的全新药物骨架的设计软件:ChemCloser。该软件通过提取先导化合物的一条长链的3D坐标获得先导化合物空间信息,并在这条长链的基础上拼接空间合理的片段,生成与原活性化合物空间形状相似的新型结构骨架。此方法不需要蛋白质的结合位点信息,只需要一个先导化合物就可以开始设计新骨架。
1 实验部分
1.1 总体设计
ChemCloser用Python 3.6语言编写,使用RDKit软件(http://www.rdkit.org)作为化学支持库,在64位Win 7系统上编译运行。ChemCloser以输入先导化合物的3D数据(pdb文件和mol文件)开始,经过获取活性化合物的3D结构和长链、将长链与片段库中的片段进行匹配获得符合要求的片段、拼接片段生成新结构等步骤,最后输出新结构的3D数据(pdb文件)。总体流程如图1所示。
ChemCloser解析活性化合物、并生成新结构的流程具体可以分为5部分:
(1) 读取活性化合物的pdb文件和mol文件,得到活性化合物的3D结构。
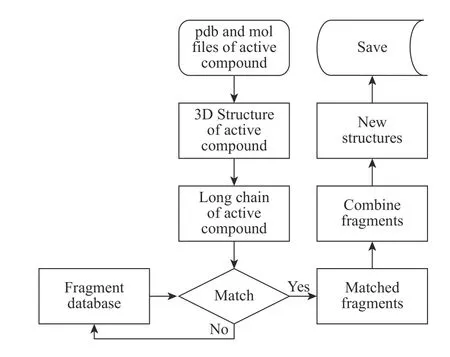
图1 ChemCloser的流程图Fig. 1 Flow chart of ChemCloser
(2)根据活性化合物的空间结构、原子间的连接关系,获得指定的两个点之间的所有路径,并提取其中一条路径(默认提取最长路径,也可以人工指定路径)作为长链。
(3)将片段库中的片段与长链进行匹配。保持长链不动,平移和旋转片段,使片段与长链尽量接近。片段与长链重叠(两个点之间的距离 < 0.1×10−10m就认为是重叠)的原子数大于3则认为匹配成功。用户可以设置需要使用的片段库、在匹配时是否要匹配片段和长链的杂化方式等超参数。匹配杂化方式会导致得到的片段数变少,但准确率提高。
(4) 将长链和匹配成功的片段进行组合并产生新结构。ChemCloser提供了两种组合拼接方式,默认的方式为:从片段库中取2 ~ 3个片段与长链进行拼接,就可以得到新结构。
(5)把新结构的3D坐标、元素符号等信息保存到pdb文件中。
1.2 片段库
ChemCloser采用片段拼接算法生成新分子,这里的“片段”指的是5 ~ 6元的单环、8 ~ 10元的并环、5 ~ 7元的桥环、10 ~ 12元的螺环等结构。ChemCloser利用RDKit软件生成6个不同规模的片段 库,这 些 片 段 库 分 别 有101、5.5×102、8.4×102、3.2×104、1.36×105、3.93×105个结构。这些基本片段最多可包含C、H、O、N、S等5种元素,其中H元素不显示标注,其余元素用原子的3D坐标、元素符号、原子杂化方式等内容表示。本软件一般采用含有550个结构式的片段库进行匹配、拼接。
1.3 实验方法
化合物1[15]是DPP-IV抑制剂(二肽基肽酶-4抑制剂)的先导化合物(半抑制浓度IC50= 5.41 μmol/L)。以化合物1作为种子分子为例给出ChemCloser的具体工作流程(图2):从化合物1中提取长链2,并在长链2的基础上拼接得到香豆素并四氢吡喃结构3、萘酚并四氢吡喃结构4的骨架。其中萘酚并四氢吡喃结构是一个长效DPP-IV抑制剂的核心骨架[16]。
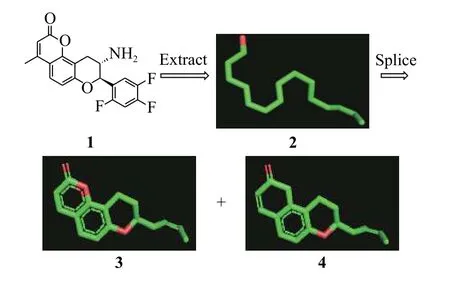
图2 提取化合物1的长链并进行片段拼接Fig. 2 Extract the long chain of compound 1 and perform fragment splicing
长链2中所有原子间的化学键均为单键,元素种类、化学键信息不会影响每个点的空间坐标; 药物化学家可以根据自己的需要,在长链2中增加或删除结点,得到链状、树状甚至环状的结构,这些结构中的点均来自于原始的化合物1,选择不同的点不影响其空间坐标。 结构式3中显然有2-萘酚结构,但是结构式显示软件PyMol将其显示为酮式。
2 结果与讨论
2.1 ChemCloser的意义
化合物1[15]作为DPP-IV抑制剂的一个先导化合物,它的稳定性、溶解性、ADME (吸收、分布、代谢和排泄)等方面可能存在问题。直接在化合物1的核心骨架(香豆素并四氢吡喃)上进行衍生或修饰可能会把这些问题带入新药中。
ChemCloser的意义:在保留化合物1的空间结构的基础上对核心骨架进行改变,为研究人员提供所有合理的核心骨架。这些骨架中可能包含完美解决上述问题的新结构,使用这些新结构可以保持原有的药效活性,并获得良好的物理化学性质。例如化合物4中的萘并四氢吡喃与化合物1的香豆素并四氢吡喃具有几乎一样的空间形状,且化合物4不含酯基,不容易被酶水解而破坏其三环结构,因此具有更好的稳定性;同时将香豆素中的酯羰基改为酚羟基(PyMol将其显示为酮式)后,为新结构提供进一步衍生和修改的可能。
2.2 软件的运行时间
ChemCloser在读取活性化合物的pdb文件、mol文件、提取活性化合物的长链等过程时运行较快,一般不超过1 s。片段与长链进行匹配过程的运行时间取决于片段库的选择和长链的原子数,一般耗时20 ~300 s。组合拼接过程是软件运行中耗时最长的部分,依据不同的化合物库和不同的长链,软件在片段拼接步骤消耗的时间变化较大,时间范围为10 min ~10 d,甚至更长。例如,用550个片段库拼接2次大约需要10 min,按照这个速度估计,用3.93×105个结构的片段库拼接2次大约需要3 540 d。
2.3 ChemCloser的性能
ChemCloser采用含有550个片段的片段库与长链2进行匹配,一共匹配成功881个状态。每一轮拼接都需要进行881次排列组合。第n轮需要881n次排列组合。显然,排列组合的次数取决于片段库的大小、片段库中的片段与长链的匹配程度。如果修改超参数,使用其他较大的片段库或者不要求匹配原子的杂化方式,则匹配成功的片段状态将明显增加,ChemCloser的排列组合性能会明显降低。基于计算机的计算性能,目前只能使用550个片段的片段库,且只能拼接2个片段。
经过两轮片段拼接、并删除价键不合理的结构,共剩余28 265个结构。用SHAFTS (Shape Feature Similarity)[17]对ChemCloser生成的结构与化合物1进行相似性打分。结果见表1。
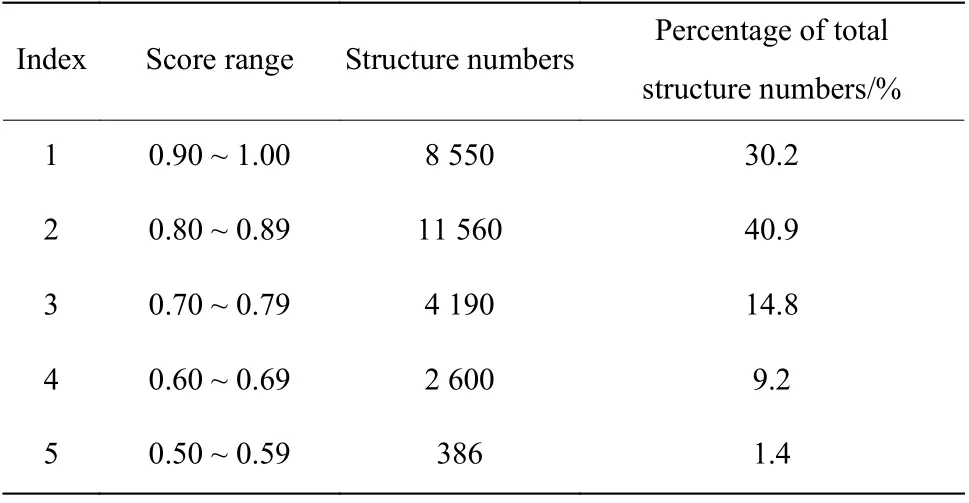
表1 ChemCloser生成的新结构与化合物1的结构相似性Table 1 Similarity between the new structure generated by ChemCloser and the structure of compound 1
从表1可以得出,有71.1%的新结构与化合物1的形状得分在0.80 ~ 1.00之间,说明ChemCloser产生的结构可以较好地保留原活性化合物的空间形状,预期它们在添加药效基团之后能与原活性化合物产生相似的药效活性。
2.4 性能的影响因素
ChemCloser采用片段与原活性化合物的长链进行匹配的方式产生新结构,总体的空间形状被长链严格限定,所以用ChemCloser产生的新结构可以得到较高的形状相似性得分。如果选取的活性化合物较大,长链较长,则片段覆盖长链的部分比较少,产生的新结构的相似性会有所降低。另一个因素是活性化合物的长链的选择。选择不同的起点、终点、不同的路径都会导致ChemCloser得到不同的长链,进而得到不同的新结构,这些新结构也会影响形状相似性得分。
2.5 结构式的多样性
相比LigBuilder软件[5]只使用57个片段就可以进行新药设计,ChemCloser使用包含550个片段的片段库,在这个规模下进行排列组合显然可以得到较好的多样性。但是ChemCloser产生的结构无法跳出长链2的限制,因此几乎无法生成其他类似形状的结构。另外,正如前面提到的,同一个活性化合物选择不同的起点、终点和路径会导致产生不同的新结构,因此只基于某一条路径生成新结构会导致多样性降低。如果在多条路径的基础上进行计算,就可以获得更多的结构式和多样性。
2.6 在人鼻病毒外壳蛋白抑制剂、人类间变性淋巴瘤激酶抑制剂方面的应用
化合物5[18]是一个人鼻病毒外壳蛋白抑制剂(图3(a))、化合物6[19]是一个人类间变性淋巴瘤激酶抑制剂(图3(b))。ChemCloser在它们的基础上生成一系列新结构。这些新结构与原活性化合物的结构相似性对比见表2。
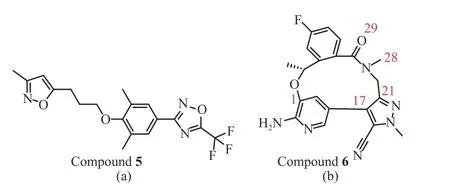
图3 化合物5(a)和化合物6(b)的化学结构式Fig. 3 Chemical structure of compound 5(a) and compound 6(b)
从表2可以得出,有63.90%的新生成的结构与化合物5有较好的形状相似性(得分0.80 ~ 1.00),说明在长链限制下生成的新结构确实能较好地保持原化合物的空间结构。
化合物6是一个环状结构,选择不同的起点和终点会得到截然不同的路径,进而生成不同的结构。例如当起点和终点分别为28和29时,生成的结构中有99.4%与化合物6有较好的形状相似性。而起点和终点分别为1和28时,没有得分为0.80 ~1.00之间的结构式,74.0%的结构与化合物6的相似性得分为0.70 ~ 0.80之间;起点和终点分别为1和21时,仅有1.16%的结构式与化合物6的相似性得分在0.80 ~ 0.90的区间,24.3%的结构式在0.70 ~0.80之间。这3组数据说明选择不同的起点和终点(即不同的路径)对生成的结构有较大影响,也再一次印证了选择合适的起点和终点(即合适的路径)的重要性。
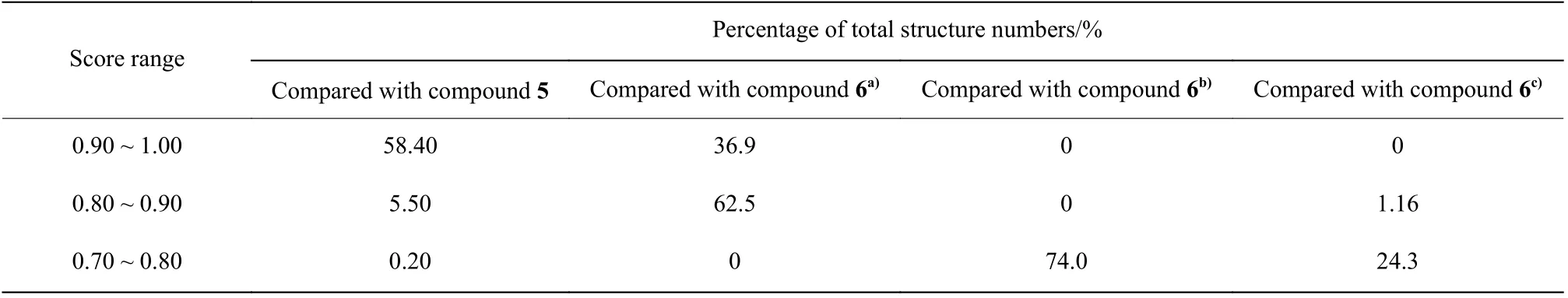
表2 基于化合物5和6生成的新结构的相似性值分布Table 2 Similarity value distribution of the new generated structures based on compound 5 and 6
2.7 存在的问题及改进方向
2.7.1 计算性能和优化方式 ChemCloser生成新结构的方式本质上是片段的排列组合。因此,当数据量较大时,一定会发生“组合爆炸”,原因是生成的中间结构无论质量好坏都进行下一轮拼接,浪费了大量CPU(Central Processing Unit)性能,由此导致ChemCloser的计算性能欠佳。这个问题将在下一个版本中解决。
2.7.2 修改原化合物的骨架对原有生物活性的影响
活性化合物的药效基团是真正产生活性的关键基团,而活性化合物的骨架只是提供安放药效基团的基本结构。当活性化合物(基本结构 + 药效基团)放到酶的结合位点时,这些药效基团可以“摆放”在合理的位置,使活性化合物与酶结合。如果活性化合物的骨架改变了,但是新骨架仍然能保持所有的药效基团“摆放”在合理的位置,那么这个新化合物的药效基团还是能与酶结合,能发挥与原化合物相同或相似的活性。ChemCloser可以保证新生成的骨架与原活性化合物有相同或相似的空间结构,且在这些骨架上连接药效基团后,这些药效基团仍然能“摆放”在合理的位置,并与酶结合。这样的化合物将会与原活性化合物具有相同或相似的生物活性。
2.7.3 新结构优劣的判断 作为药物的核心骨架,应该具有较好的稳定性、适中的刚性等物理、化学特性。如果核心骨架上存在酯基、酰胺、醛基、(半)缩醛、(半)缩酮等容易变化的基团,则该骨架很容易被降解(破坏);如果核心骨架是大的芳环,则会导致药物刚性太强,溶解性降低,导致化合物无法突破人体内的各种屏障,到达作用位点并发挥药效。我们对片段库中的基本结构进行筛选,删除化学性质不稳定、刚性太强的片段,使生成的新骨架有较好物理、化学性质。但是,即使片段库中的基本结构都是化学性质稳定、刚性适中的片段,在进行片段拼接后仍然可能生成不稳定、刚性过强的结构。目前只能人工判断新骨架的优劣,而无法让ChemCloser自动判断,这个问题需要在下一个版本中尝试解决。
2.7.4 提高生成结构质量的方式 ChemCloser生成的骨架质量差别较大,需要通过多方面对新骨架进行考察。例如用SHAFTS[17]对新骨架与原活性化合物进行空间相似性打分、查找是否存在不稳定的基团、通过引入受体的结构,计算新化合物与受体的结合数据,对比原化合物与受体的结合数据,可以进一步确定新骨架是否合理。综合应用这些方法可以在所有结构中筛选出质量最好的一个或几个骨架。
2.7.5 判断生成的化合物新颖性的方法 ChemCloser
只负责生成包括原活性化合物的骨架,以及新骨架在内的所有骨架,不负责判断新骨架是不是“新颖的”。对于ChemCloser生成的新骨架,必须要由药物设计者通过全面查阅文献、专利等资料后判断该骨架是不是具有新颖性。
2.7.6 路径的选择方式 ChemCloser是一个半经验的药物设计软件,并不能全自动地设计活性化合物的新骨架。选择不同的起点和终点,活性化合物会有多条路径,ChemCloser默认选取活性化合物中最长的一条长链。但最长的路径并不一定是(最)合理的路径。我们认为在药物设计之前,先要研究活性化合物中哪些是药效基团,哪些是辅助基团,活性化合物的关键骨架是什么,在确定这些信息之后,再进行有取舍的路径选择。合理的路径应该保证路径上能够放置所有的药效基团,并获得活性化合物的空间信息。而这样的路径可能不再是链状结构,有可能是树状结构、甚至是图(包含环)。如果依靠计算机的路径规划算法可能很难区分哪些是必须的片段,哪些是不重要的片段,由此很难获得真正合理的路径(长链)。
3 结 论
(1) 开发了一款基于片段拼接、空间结构匹配的全新药物骨架的设计软件:ChemCloser,它能提取活性化合物的一条长链的3D坐标,并在该长链的基础上拼接空间合理的片段,得到结构新颖、空间形状与活性化合物相似的分子骨架,可以为药物设计人员提供更多、更优秀的核心骨架。
(2) 以DPP-IV抑制剂的先导化合物1为设计模型,分析了ChemCloser的性能、性能影响的因素、生成结构式的多样性。考察了ChemCloser在人鼻病毒外壳蛋白抑制剂和人类间变性淋巴瘤激酶抑制剂方面的应用,新结构同样显示出较好的结构相似性,并且再一次印证了选择不同的长链对新结构有较大影响。
(3) 分析了ChemCloser存在的几个问题。ChemCloser采用排列组合的方式拼接生成新结构式,这种方式在数据量较大时会发生“组合爆炸”的问题。目前ChemCloser不能自动判断新结构的优劣,也不能判断生成的结构是不是新颖,这些都需要人工判断。分析了修改原化合物的骨架对原有生物活性的影响、提高生成结构的质量的方式、路径的选择方式等问题。
