基于基因组挖掘技术的新型谷氨酸脱羧酶基因挖掘及表达鉴定
2021-06-04解玉丽贾园园王红艳刘先吏罗湘艳唐存多
李 祥,解玉丽,贾园园,张 闪,王红艳,刘先吏,罗湘艳,唐存多,2,*
(1.南阳师范学院 昆虫生物反应器河南省工程实验室,河南 南阳 473061;2.车用生物燃料技术国家重点实验室,河南 南阳 473061)
γ-氨基丁酸(γ-aminobutyric acid,GABA)是广泛存在于自然界中的一种非蛋白质氨基酸,在医药、食品和化工行业均有较大的应用前景[1-2]。在医药行业,GABA作为一种抑制性神经递质[3],可用于调节血压和治疗精神疾病[4-6]。在食品行业,GABA可作为重要的食品添加剂,用于制备各种具有镇静安神、降压补脑功效的功能性食品[7-8]。在化工行业,可用作生物可降解塑料——尼龙4的前体物质,进行可降解塑料的合成[9-10]。目前,GABA的合成方法主要包括化学合成、植物富集和生物合成法[2,11-12]。传统的化学合成法能耗高、污染大,不利于可持续发展[13]。植物富集法又受限于原料中GABA含量较低,难以大规模生产。而生物合成法不受资源、环境和空间的限制,更绿色、更环保,它已在GABA合成领域最受青睐[1]。
谷氨酸脱羧酶(glutamate decarboxylase,GAD,EC 4.1.1.15)是磷酸吡哆醛依赖性酶,在L-谷氨酸不可逆脱羧生成GABA的过程中起关键作用[14]。GAD广泛存在于各种生物体中,许多微生物均已检测到GAD活力[15]。由于乳酸菌用于食品发酵的历史悠久,且还具备一定的保健潜力,因此许多有关GABA和GAD的研究都聚焦于乳酸菌,主要包括短乳杆菌(Lactobacillus brevis)[16]、副干酪乳杆菌(L. paracasei)[17]、鼠李糖乳杆菌(L. rhamnosus)[18]、清酒乳杆菌(L. sakei)[19]、植物乳杆菌(L. plantarum)[20]、酵素乳杆菌(L. zymae)[21]、发酵乳杆菌(L. fermentum)[15]和乳酸乳球菌(Lactococcus lactis)[22]。然而,受限于乳酸菌生长速度缓慢,难以实现高密度发酵,因此从乳酸菌中提取GAD难以满足GABA的大规模生产需求[15]。为了获得足够的GAD,以满足GABA工业生产的需求,挖掘乳酸菌来源的GAD并实现在大肠杆菌(Escherichia coli)中高效异源表达受到了广泛关注[15,23]。迄今为止,尽管已有许多关于GABA和GAD的研究报道,但它们产业化程度仍不高,大多数都停留在实验室水平的研究阶段。这主要归因于GAD的催化活性低、稳定性和pH值适应性差,以及生物催化法制备GABA酶的使用成本太高。如何获得催化活性高、稳定性好的优良GAD已成为企业和科研人员当前所面临的巨大挑战,也是降低生物催化法制备GABA生产成本所必须突破的瓶颈[24-25]。
自然界中蕴藏着丰富的未被人工培养微生物及未被发掘新型酶资源,发掘这些未知的酶类并加以适当改造以满足工业化生产的需求,吸引了广大科研工作者的关注[26]。随着高通量测序技术的不断发展,基因组序列数据也日益丰富,为新酶的挖掘提供了丰富的基因资源[27-28]。基因组挖掘技术可以绕过传统的微生物分离筛选及蛋白的分离纯化等过程,实现从基因组数据库到真实酶数据库的跨越,进一步丰富了可被利用或改造的酶资源[29-30]。
本研究拟借助基因组挖掘技术,从公认安全的(generally recognized as safe,GRAS)有益细菌的基因组中挖掘新型的GAD基因,利用E. coli作为宿主,构建能够高效表达重组GAD基因工程菌,并进行全细胞催化L-谷氨酸合成GABA的研究,旨在为实现GABA高效、廉价的绿色生物合成奠定理论基础。
1 材料与方法
1.1 材料与试剂
1.1.1 试剂
L-谷氨酸、磷酸吡哆醛、硼酸、苯酚、次氯酸钠、GABA标准品 北京索莱宝科技有限公司;三乙胺、苯异硫氰酸酯(phenylisothiocyanate,PITC) 麦克林化学试剂有限公司;异丙基-β-D-硫代半乳糖苷(isopropyl-β-D-thiogalactoside,IPTG) 生工生物工程(上海)股份有限公司;甲醇(色谱纯) 天津科密欧化学试剂有限公司;一站式His标记蛋白质微量纯化套装 北京天恩泽基因科技有限公司;其他试剂均为国产或进口分析纯。
1.1.2 菌株、质粒与培养基
携带原核表达质粒的E. coliBL21/pET28a菌株由本课题组保藏[9],主要用作GABA生物合成反应的阴性对照。
LB培养基:10 g/L蛋白胨、10 g/L氯化钠和5 g/L酵母提取物,制作固体平板时添加15 g/L琼脂粉,自然pH值,121 ℃灭菌20 min,用于E. coli的培养。
1.2 仪器与设备
PowerPac™ HC/Mini-PROTEAN®蛋白电泳系统、UNIVERSAL Hood凝胶成像系统 美国Bio-Rad公司;Hypersil C18柱 美国Thermo Scientific公司;UV1000紫外-可见分光光度计 上海天美科学仪器有限公司。
1.3 方法
1.3.1 新型GAD的基因挖掘
以研究比较透彻、催化活性较高的L. brevis来源GAD(LbGAD)蛋白序列为探针[31-32],以GRAS有益细菌(主要包括乳酸菌、肠球菌)的基因组序列为搜索对象进行BLAST分析,从搜索到的序列中找出一系列基因组信息来源且未经表达鉴定、假定的GAD蛋白。然后,对这些潜在的GAD蛋白序列利用ClustalX2和MEGA6.0软件进行多序列比对和进化树的构建。基于对进化树的分析,选定几个代表性的序列借助Optimizer服务器以E. coliK12菌株的密码子使用频率表为依据,进行密码子优化。经过对目标基因和表达质粒多克隆位点的酶切位点分析后,在目标基因的上下游分别加上合适的酶切位点,最后委托苏州泓迅生物科技有限公司进行全基因的合成。
1.3.2 重组E. coli的诱导表达
采用低温、低诱导剂浓度的诱导策略进行[33]。分别将E. coliBL21/pET28a和携带不同GAD编码基因的重组E. coli单菌落接种至4 mL含50 μg/mL卡拉霉素的LB液体培养基中,37 ℃、200 r/min条件下振荡培养14 h,以2%接种量转接至100 mL新鲜LB液体培养基中,37 ℃、200 r/min再培养约2.5 h,加入1 mol/L IPTG溶液至终浓度为0.1 mmol/L,16 ℃、200 r/min诱导培养20 h;取10 mL菌液于8 000 r/min、4 ℃离心5 min,收集菌体,将菌体重悬后进行超声破碎,并进行十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis,SDS-PAGE)分析[33]。重组酶的纯化按照一站式His标记蛋白质微量纯化套装试剂说明书进行。
1.3.3 酶活力分析
由于PITC衍生化所需的时间较长[34],不利于GABA的快速定量,因此本研究中重组GAD活力的快速测定参照比色法[35-36]略作修改。酶活力测定的反应体系为1 mL,包含0.01 mmol/L磷酸吡哆醛、20 mmol/LL-谷氨酸、200 mmol/L pH 5.0的Na2HPO4-柠檬酸缓冲液,37 ℃保温2 min后加入100 μL稀释适当倍数的酶液,迅速混匀后反应5 min,迅速放入沸水浴终止反应。取400 μL反应液,依次加入400 μL 0.2 mol/L pH 9.0的硼酸缓冲液、1.0 mL 6%苯酚和1.0 mL次氯酸钠溶液(有效氯大于7.5%),充分振荡后,沸水浴中反应10 min迅速在冰浴中冷却显色20 min,待呈现蓝绿色后加入2.0 mL 60%乙醇溶液,最后在643.5 nm波长处测定吸光度,以底物溶液中加入等量蒸馏水为对照组。由此快速测定反应生成GABA的量,以测定GAD活力。在测定条件下每分钟生成1 μmol GABA所需的酶量定义为1 个酶活力单位(U)。
1.3.4 重组酶的温度特性分析
参照1.3.3节方法,分别在30~60 ℃(以5 ℃为间隔)测定各重组酶的催化活性以获得最适反应温度;将各重组酶分别在20~60 ℃(以5 ℃为间隔)保温1 h,然后在各自最适反应温度下测定各自的残留酶活力,以冰浴保存1 h后的残余酶活力为100%,以此考察各重组酶的热稳定性。
1.3.5 重组酶的pH值特性分析
参照1.3.3节方法,在各自最适反应温度下,分别在pH 2.0~6.0(以0.5为间隔)下测定各重组酶的催化活性以获得最适反应pH值;将各重组酶分别在pH 2.0~6.0(以0.5为间隔)缓冲液中冰浴1 h,然后在各自最适反应温度和最适反应pH值下测定各自的残留酶活力,以未经处理为100%,以此考察各重组酶的pH值稳定性。
1.3.6 重组酶的动力学参数
将辅酶磷酸吡哆醛的终浓度定为0.01 mmol/L,逐步提高底物的终浓度,测定各酶在不同底物浓度下的反应速率,利用Origin 9.0软件进行非线性拟合,得出各酶对底物的Km、Kcat和Vmax值。
1.3.7 重组E. coli全细胞转化L-谷氨酸合成GABA
为了提高GABA生产的时空得率,本研究采用边诱导边转化的“双边工艺”方式进行。参照1.3.2节条件分别对E. coliBL21/pET28a和携带不同GAD编码基因的重组E. coli进行前期的种子培养,加入1 mol/L IPTG溶液至终浓度为0.1 mmol/L后,紧接着添加L-谷氨酸至终质量浓度为6 g/L,添加磷酸吡哆醛的母液至终浓度为0.5 mmol/L,先在16 ℃、200 r/min条件下培养24 h,然后在30 ℃、200 r/min条件下培养24 h,期间每4 h取样检测GABA生成量。
1.3.8 GABA的高效液相色谱(high performance liquid chromatography,HPLC)分析
GABA的精确定量采用柱前PITC衍生HPLC进行[34],称取GABA标准品溶解于超纯水中,取不同体积的标准品溶液加入到10 mL的离心管中,制成不同浓度梯度的GABA标准液,然后分别加入500 μL 0.1 mol/L PITC-乙腈、500 μL 1 mol/L三乙胺-乙腈,充分混匀后室温放置反应1 h,反应结束后加入3 mL 50%乙腈溶液,混合均匀,再加入5 mL正己烷,混合均匀后静置10 min,取下层溶液,用0.22 μm滤膜过滤,取20 μL滤液进行HPLC分析,获得各浓度GABA峰面积后,进行线性拟合获得回归方程及相关系数。HPLC色谱柱为Thermo Hypersil C18柱,检测波长254 nm,柱温25 ℃,流动相为体积分数10%的甲醇溶液,流速1 mL/min。采用同样的方法对转化产物和L-谷氨酸分别进行衍生化和HPLC分析,根据产物的保留时间进行定性分析,同时根据测得产物峰面积,由获得的回归方程进行定量分析。
1.3.9 常用网站及软件
NCBI(http://www.ncbi.nlm. nih.gov/)数据库和Basic Local Alignment Search Tool(https://blast.ncbi.nlm.nih.gov/Blast.cgi):用于蛋白质、基因序列及蛋白质3-D结构的搜索;Optimizer(http://genomes.urv.cat/OPTIMIZER/):用于密码子的优化;ClustalX2:用于蛋白质的多序列比对;MEGA6.0:用于进化树的构建;DNAMAN:用于基因序列及限制性酶切位点分析。
1.4 数据统计及图表绘制
图片均用Adobe Photoshop CS 8.0软件进行色阶、对比度调节及标注等处理。简单的数据处理及分析均借助Origin 9.0进行。
2 结果与分析
2.1 GAD的基因挖掘

图1 探针及潜在的GAD进化树Fig.1 Phylogenetic tree for the probe and putative glutamate decarboxylases
如图1所示,以在进化树上的距离为依据选取了4 个有代表性的序列,包括L. lactisCICC20209、E. sulfureus、L. senmaizukei和L. brevisATCC 367来源的4 个潜在的GAD,并将其分别命名为LlGAD、EsGAD、LsGAD和LbGAD。其中,LsGAD与已报道的GAD相似度最高为82.01%(序列号为BAN05709.1)[31],而EsGAD与报道的GAD相似度最高为70.18%(序列号为KRN72673)[37],相似度表明这2 个酶在基因序列上均具有一定的新颖性。对选定的4 个基因进行了密码子优化,然后对目标基因和pET28a多克隆位点的酶切位点进行分析,在目标基因上下游分别加上BamH I和XhoI酶切位点后,委托苏州泓迅生物科技有限公司进行全基因合成,并克隆至pET28a上,构建出一系列携带潜在GAD编码基因的重组E. coli,分别命名为E. coliBL21/pET28a-LsGAD、E. coliBL21/pET28a-LlGAD、E. coliBL21/pET28a-EsGAD和E. coliBL21/pET28a-LbGAD。
2.2 重组E. coli的诱导表达及纯化
分别将E. coliBL21/pET28a、E. coliBL21/pET28a-LsGAD、E. coliBL21/pET28a-LlGAD、E. coliBL21/pET28a-EsGAD和E. coliBL21/pET28a-LbGAD按照1.3.2节方法进行诱导表达。将100 mL发酵液中的菌体离心收集,然后超声破碎,高速离心20 min后收集裂解上清液,将上清液过0.45 μm滤膜后用一站式His标记蛋白质微量纯化套装进行纯化,并用10 kDa截留分子质量的超滤离心管进行咪唑去除和产物浓缩,最终分别将各重组蛋白定容至1.5 mL。将裂解上清液和纯化后产物分别进行SDS-PAGE分析,结果如图2所示,E. coliBL21/pET28a-EsGAD、E. coliBL21/pET28a-LbGAD、E. coliBL21/pET28a-LsGAD和E. coliBL21/pET28a-LlGAD的裂解上清液在约53 kDa处有明显的特异性条带,与理论分子质量一致,表明这4 个潜在的GAD均成功实现了在E. coli中的可溶性表达,其中E. coliBL21/pET28a-EsGAD的蛋白可溶性表达水平最差。纯化产物均在约53 kDa处出现了单一条带,表明经过亲和层析获得了电泳纯的4 个重组酶,可以用于后续酶学特性的分析。
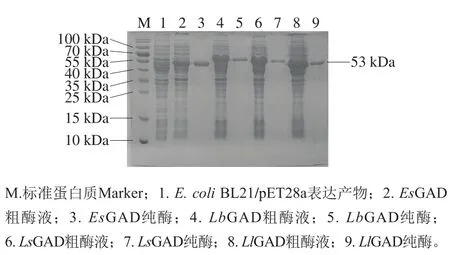
图2 代表性重组GAD的SDS-PAGE分析Fig.2 SDS-PAGE analysis of representative recombinant glutamate decarboxylases
酶活力测定结果显示,E. coliBL21/pET28a-EsGAD、E. coliBL21/pET28a-LbGAD、E. coliBL21/pET28a-LsGAD和E. coliBL21/pET28a-LlGAD发酵液中GAD活力分别为8.38、28.97、34.17 U/mL和38.91 U/mL,LsGAD和LlGAD在E. coli中的表达水平明显高于探针LbGAD的表达水平,这表明在催化L-谷氨酸生成GABA的反应中前两者可能会有更高的效率。经纯化后,reEsGAD、reLbGAD、reLsGAD和reLlGAD对L-谷氨酸的比活力分别为114.35、108.63、139.60 U/mg和46.62 U/mg,其中reLsGAD比活力最高,约为探针(reLbGAD)的1.4 倍。
2.3 重组GAD的温度特性
由图3A可知,LsGAD的最适反应温度最低,仅为40 ℃,它更接近于菌体的最适生长温度。结果表明在生理条件下,LsGAD较其他GAD可能会更具催化潜力。如图3B所示,4 个重组GAD的热稳定性均欠佳,游离酶在40 ℃保温1 h后残留酶活力均只有50%左右,这会限制它在GABA生物合成中的应用,在今后研究中有必要通过固定化、定向进化或理性设计等手段对其热稳定性进行改造,以满足工业化生产的需求[38-39]。在本研究中,将采取全细胞催化的方法进行GABA的生物合成,以提高GAD在反应条件下的稳定性。

图3 重组GAD的最适温度(A)和热稳定性(B)Fig.3 Optimal temperature (A) and temperature stability (B) of recombinant glutamate decarboxylases
2.4 重组GAD的pH值特性

图4 重组GAD的最适pH值(A)和pH值稳定性(B)Fig.4 Optimal pH (A) and pH stability (B) of recombinant glutamate decarboxylases
由图4A可知,4 个重组GAD均为嗜酸性酶,它们的最适pH值在4.0~5.5之间,其中LsGAD的pH值适应性最广,在4.5~6.0之间均能保持80%以上的相对酶活力,具有更大的应用潜力。由图4B可知,4 个重组GAD对pH值较为敏感,当稍微偏离其最适pH值时,其稳定性明显下降,因此在使用过程中一定要严格控制反应体系的pH值。
2.5 重组GAD的动力学参数分析

表1 重组GAD对L-谷氨酸的动力学参数Table 1 Kinetic parameters for the activity of recombinant glutamate decarboxylases toward L-glutamic acid
表1结果显示,与探针及其他2 个重组GAD相比,LsGAD对L-谷氨酸的Km值最低,仅为3.37 mmol/L,表明它对L-谷氨酸具有更高的亲和力。而LsGAD的Kcat/Km值为10.27 L/(mmol·s),也明显高于探针及其他2 个重组谷氨酸脱氢酶,这表明LsGAD较其他几个GAD可能会具有更高催化效率。
2.6 GABA的生物合成
采用1.3.7节边诱导边转化的“双边工艺”对L-谷氨酸进行全细胞催化的生物转化,将反应产物按1.3.8节方法进行HPLC检测,在保留时间为2.94 min和2.23 min处均有明显的特征峰,表明已有部分L-谷氨酸(2.23 min)转化为GABA(2.94 min)。6 g/L的L-谷氨酸在上述转化工艺下的反应进程曲线如图5所示,在前20 h的诱导时间段,4 个携带GAD基因的工程菌均能产生一定的GABA,其得率约为10%,再经过24 h的诱导后,GABA得率最高可达58%,最低约为40%,离理论得率仍有一定的差距,在后续研究中仍需对转化条件进行系统的优化。但相比传统的先诱导后转化的分步工艺,该双边工艺节约了一定的转化时间,提高了GABA的时空生产效率[15]。

图5 不同E. coli工程菌催化下的GABA反应进程曲线Fig.5 Time course curves of GABA production catalyzed by engineered E. coli cells
3 讨 论
随着高通量测序技术的不断发展,基因组数据在不断丰富,迄今为止已有222 395 个原核生物的基因组序列被先后报道,且仍在呈递增趋势。海量基因组序列中包含了大量假定酶基因,为新酶的发掘提供了丰富资源[26]。采用基因组挖掘技术,可以快速地将假定酶变为真实酶,为生物催化提供更多的选择[27]。
本研究采用基因组挖掘技术,以LbGAD的蛋白序列为探针,从乳酸菌属和肠球菌属的基因组中挖掘到了3 个假定的GAD(LsGAD、EsGAD和LlGAD),其中LsGAD与已报道的序列相似度最高为82.01%[31],而EsGAD与已报道的序列相似度最高为70.18%[37],均具有较高的新颖性。借助pET28a质粒将4 个基因实现了在E. coliBL21中的可溶性表达,E. coliBL21/pET28a-EsGAD、E. coliBL21/pET28a-LbGAD、E. coliBL21/pET28a-LsGAD和E. coliBL21/pET28a-LlGAD发酵液中GAD活力分别为8.38、28.97、34.17 U/mL和38.91 U/mL,LsGAD和LlGAD在E. coli中的表达水平明显高于探针LbGAD的表达水平,表明其全细胞催化L-谷氨酸合成GABA的应用中可能更具优势。此外,reLsGAD对L-谷氨酸的酶活力最高,可达139.60 U/mg,约为探针(reLbGAD)的1.4 倍。而reLlGAD对L-谷氨酸的酶活力最低,仅为46.62 U/mg,但是其可溶性的蛋白表达含量很高(图3),所以体现出的酶活力表达水平仍然最高。
温度特性结果显示,LsGAD的最适反应温度最低,仅为40 ℃,更接近于菌体的最适生长温度,表明在生理条件下,LsGAD较其他GAD可能会更具催化潜力。pH值特性结果表明,4 个重组GAD均为嗜酸性酶,它们的最适pH值在4.0~5.5之间,其中LsGAD的pH值适应性最广,在4.5~6.0之间均能保持80%以上的相对酶活力,可能会具有更大的应用潜力。此外,动力学参数结果显示,LsGAD的Kcat/Km值为10.27 L/(mmol·s),也明显高于探针及其他2 个重组GAD。从这些酶学特性的参数比较分析看,均体现出LsGAD的优越性,下一步选择其做进一步研究,包括采用共表达质粒提高基因的拷贝数及在食品级乳酸菌表达系统中进行表达等。
最后,采用边诱导边转化的“双边工艺”对L-谷氨酸进行全细胞催化的生物转化,6 g/L的L-谷氨酸经过24 h转化后,GABA得率最高可达58%。相比传统的先诱导后转化分步工艺,该双边工艺节约了一定的转化时间,提高了GABA的时空生产效率[15]。总之,本研究实现了GAD从基因组数据到真实酶的跨越,获得了数个性能优良的GAD,初步实现了GABA的生物合成,为实现GABA低成本、规模化的生物合成提供一定的参考。
