Ni,Pd,Pt负载α-MoC催化水煤气变换反应理论
2021-05-21邹雪燕左志军
史 肖,邹雪燕,黄 伟,左志军
(1.太原理工大学 省部共建煤基能源清洁高效利用国家重点实验室,山西 太原 030024; 2.太原理工大学 煤科学与技术教育部重点实验室,山西 太原 030024)
水煤气变换反应(WGS,CO(g)+H2O(g)→CO2(g)+H2(g))是CO去除和H2制备的途径[1],也是合成气转化过程中的重要反应,如甲醇合成[2]和费托合成[3]。常见的WGS反应催化剂有Au,Cu,Pd,Ni和Pt基等催化剂[4-8]。为了提高催化剂的活性,通常通过添加助剂[9-12]或改变载体[13-15]的方法。
过渡金属碳化物是一种金属间填充型化合物,是由碳原子填隙式融进过渡性金属的晶格中形成。由于其表面性质和催化活性类似于Pt等贵金属,目前广泛应用于催化加氢、脱氢、WGS和异构化等反应[16-17]。如,Au/α-MoC用于WGS反应时,反应速率和CO转化率分别是Cu/Zn/Al2O3催化剂的354倍和17.8倍[18],同时Au/α-MoC催化剂的活性明显优于Au/β-MoC。α-MoC对WGS反应起到了重要的作用,因此,笔者选取了α-MoC作为载体。
理论计算结果显示,α-MoC(111)载体对WGS反应没有催化效果,但是能够促进H2O的解离。当其负载Au和Cu后能够提高WGS反应在2种催化剂表面的活性[18-19]。Pd,Ni,Pt基催化剂也是WGS反应的常用催化剂,为了进一步了解α-MoC载体在WGS反应中的作用和WGS反应机理,笔者使用密度泛函理论(DFT)和动力学蒙特卡罗(KMC)方法系统的研究这3种金属负载α-MoC催化剂上的WGS反应过程,研究不同反应温度时的反应速率和催化转换频率(TOF),并对WGS反应催化剂的优化提供指导。
1 计算方法和模型
计算使用VASP软件[20-22],采用了GGA-PBE泛函求解电子交换相关能[23]。平面波截断能取值为 415 eV,布里渊区的k点选择 3×3×1[24]。在结构优化的过程中当力的变化小于0.1 eV/nm和能量的变化小于1.0×105eV/atom时达到收敛标准。采用CI-NEB方法进行过渡态搜索,对达到收敛标准的过渡态进行频率分析,通过唯一虚频确定过渡态[25]。
首先对金属晶胞M(M=Ni,Pd,Pt)和α-MoC晶胞进行了优化,优化后对应的的晶格常数分别为aNi=0.351 3 nm,aPd=0.393 7 nm,aPt=0.396 7 nm,aα-MoC=0.437 0 nm,分别与实验值aNi=0.352 4 nm,aPd=0.389 1 nm,aPt=0.392 4 nm和aMoC=0.427 8 nm接近[26-27]。因此,选择的计算方法和参数对本文的计算体系是合理的。α-MoC(111)采用7层3×3的平板模型,1.5 nm的真空层。由于H2O的快速解离,α-MoC表面的Mo位点被O占据[19],如图1所示,图1中top,hol,bri为吸附位点。计算过程中固定底面两层,其余原子弛豫。金属M4簇负载存在2种构型:平面簇构型(2d rhombicM4)和四面体簇(3d-tetrahedralM4),对应的吸附能分别为Ni4(2d:-8.41 eV;3d:-7.63 eV),Pd4(2d:-8.56 eV;3d:-6.77 eV),Pt4(2d:-10.20 eV;3d:-9.32 eV)。计算结果表明,平面簇构型更稳定,故仅考虑了2d-M4负载在α-MoC(111)表面。
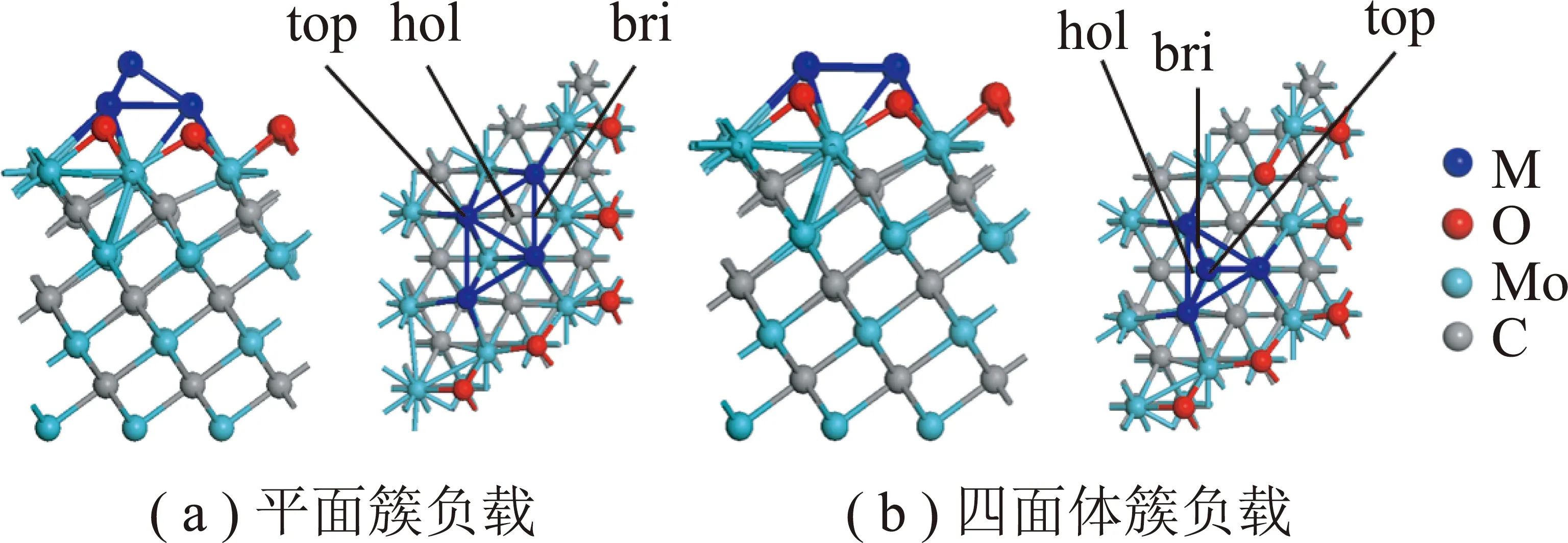
图1 M4/α-MoC(111)的侧视图和俯视图Fig.1 Top and side view of M4/α-MoC(111)
2 结果与讨论
对于水煤气变换反应,反应机理主要有2种:① 氧化还原机理:CO直接与H2O分解产生的氧结合,生成CO2;② 甲酸盐途径或者羧酸盐途径:CO与H2O分解产生的羟基结合,生成甲酸盐或羧酸盐中间体,最后分解成CO2[28-30]。反应过程中所涉及到的中间体,反应物和产物的最稳定吸附构型如图2所示。
图2和表1显示,反应物、中间体和产物在3类催化剂表面的最优吸附位相近,只有*H在Pt4/α-MoC(111)和*CO2在Ni4/α-MoC(111)的吸附例外。对中间体来说(*O,*OH,*CO2,*H2O,*CHOO,*COOH),它们在Pd4/α-MoC(111)和Pt4/α-MoC(111)的吸附能相近,吸附稳定性低于对应物种吸附在Ni4/α-MoC(111)。
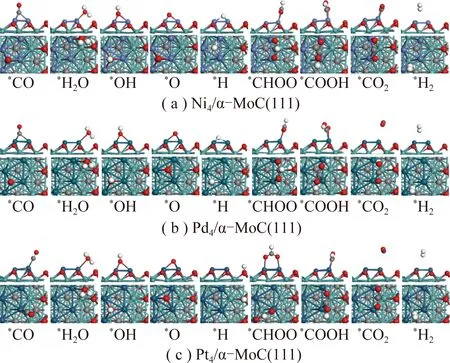
图2 WGS反应所涉及的各中间体在Ni4/α-MoC(111)的稳定吸附构型Fig.2 Adsorption configurations of intermediates involved in the WGS on the surfaces of Ni4/α-MoC(111)
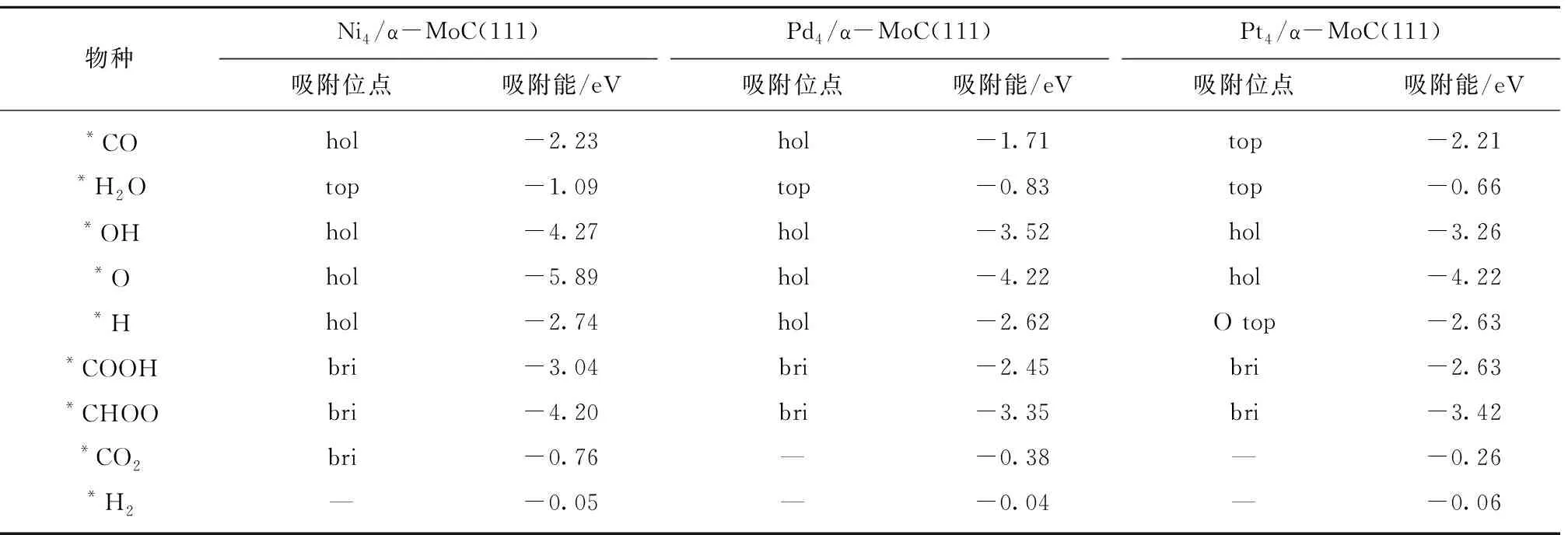
表1 M4/α-MoC(111)面WGS反应过程中涉及到的中间体,反应物和产物的最优吸附位和吸附能Table 1 Adsorption energy and adsorption site of the possible intermediates,reactants and productions during the WGS reaction on M4/α-MoC(111)
反应物*CO吸附在Ni4/α-MoC(111)和Pd4/α-MoC(111)的吸附能相近(-2.23和-2.21 eV),而在Pt4/α-MoC(111)面的吸附稳定性低于Ni4/α-MoC(111)和Pd4/α-MoC(111)面。相比CO在Ni(111),Pd(111)和Pt(111)的吸附能,载体α-MoC提高了CO在Ni和Pd的吸附稳定性,但是降低了在Pt上的吸附稳定性[31-32]。对*H2O来说,载体α-MoC一定程度上提高了其在Ni,Pd和Pt表面的吸附稳定性[31-32]。
优化后,产物*CO2远离Pd4/α-MoC(111)和Pt4/α-MoC(111)面,但是其吸附在Ni4/α-MoC(111)的bri位,这是由于*CO2在Ni4/α-MoC(111)面的吸附稳定性强于Pd4/α-MoC(111)和Pt4/α-MoC(111)面。总体来说,相较Ni(111),Pd(111)和Pt(111),α-MoC(111)提高了CO2在3种催化剂表面的吸附稳定性[31-32]。对于H2来说,其在3个表面的吸附能力很弱,载体对其吸附基本没有影响。
*H2O的解离(*H2O+*→*OH+*H)是WGS反应的第1步,在Ni4/α-MoC(111)上*H2O解离的能垒为1.16 eV。对于氧化还原机理,*O生成存在2种方式:①*OH直接裂解(*OH+*→*O+*H)能垒Ea为0.95 eV;②*OH歧化反应(*OH+*OH→*H2O+*O)能垒为0.37 eV。最后,*CO氧化生成*CO2需克服1.62 eV的能垒。对于甲酸盐和羧酸盐路径,*CO与*OH反应,生成*CHOO(Ea=2.84 eV)或*COOH(Ea=1.61 eV)。结果表明,在Ni4/α-MoC(111)上氧化还原机理容易发生,速控步骤为*CO氧化。
在Pd4/α-MoC(111)上,*H2O克服1.23 eV能垒解离生成*OH 和*H。当发生氧化还原反应时,*OH歧化反应的能垒为0.35 eV,而*OH直接解离的能垒为1.02 eV。最后,*CO氧化生成*CO2的能垒为0.64 eV。在发生甲酸盐或羧酸盐路径中,*CO与*OH反应生成*CHOO和*COOH的能垒分别为1.64和1.28 eV。因此,在Pd4/α-MoC(111)上,WGS反应的氧化路径为主要路径,H2O的解离为速控步骤。
在Pt4/α-MoC(111)上,H2O的解离能垒为0.86 eV。当发生氧化还原反应时,*OH+*→*O+*H和*OH+*OH→*H2O+*O的反应能垒分别为1.28和0.35 eV,*CO氧化生成*CO2的能垒为0.81 eV。在发生甲酸盐或羧酸盐路径中,*CO与*OH反应生成*CHOO和*COOH的能垒分别为1.72和0.66 eV。由于生成*COOH的能垒远远低于生成*HCOO的能垒,因此*CO和*OH反应优先生成*COOH。最后,经*COOH与*OH反应生成*CO2和*H2O(Ea=0.42 eV)。
基于此,根据WGS反应的正逆反应能垒和不同温度时的指前因子,利用KMC研究了WGS反应在Ni4/α-MoC(111),Pd4/α-MoC(111)和Pt4/α-MoC(111)上的反应路径和催化转换频率。对于反应物和产物的吸附和脱附,考虑了熵的贡献[33]。KMC模型和计算参数等详细信息见文献[34]。
KMC结果显示,标准大气压(n(CO)∶n(H2O)=1∶1,物质的量之比)和反应温度为500 K时,在Ni4/α-MoC(111)和Pd4/α-MoC(111)催化剂上,水煤气变换的反应机理为氧化还原机理如图3所示,图3中,TS为过渡态。即CO(g)+H2O(g)→*CO+*H2O→*CO+*OH+*H→*CO+*O+*2H→*CO2+*H2→CO2(g)+H2(g)。需要指出的是,尽管*OH+*OH→*H2O+*O的反应能垒远远小于*OH+*→*O+*H的反应能垒,然而由于高的*H2O→*OH+*H反应能垒导致表面*OH覆盖度过低。因此,KMC结果显示*OH的直接解离是*O产生的反应途径。在Pt4/α-MoC(111)催化剂上,水煤气变换的反应路径为羧酸盐路径如图4所示,即CO(g)+2H2O(g)→*CO+2*H2O→*CO+2*OH+2*H→*COOH+*OH+2*H→*CO2+*H2O+*H2→CO2(g)+H2O(g)+H2(g)。由于H2和CO2的TOF相等,因此图5仅列出了H2的TOF(反应条件为一个大气压下,CO与H2O物质的量之比为1∶1)。总体来说,H2的TOF随反应温度的升高而升高。H2在Ni4/α-MoC(111)和Pd4/α-MoC(111)的TOF偏低。在500 K时催化剂单位活性位点上转换次数约为0和0.056 s-1,这是由于WGS反应过程中反应能垒较高造成的。对于Pt4/α-MoC(111),值得注意的是,400 K时,H2在催化剂单位活性位点上转换次数约为0,这是因为Pt4/α-MoC(111)上CO的脱附能较高(1.07 eV),活性位点被CO所覆盖。随着反应温度的升高,CO的脱附能(500 K下脱附能为0.83 eV)降低,H2在催化剂单位活性位点上的TOF约为6.3 s-1,WGS反应发生。结果表明,*CO的强稳定性不利于WGS反应的进行。当反应温度为500 K时,H2的TOF的排序是:Pt4/α-MoC(111)≫Pd4/α-MoC(111)>Ni4/α-MoC(111)。因此,当3种金属负载在α-MoC载体上时,Pt4/α-MoC(111)催化剂活性最好。同时,其反应活性高于Pt/Al2O3(TOF≈0.7 s-1)和Pt/TiO2(TOF≈0.2 s-1)[35],这表明Pt/α-MoC是WGS反应的高活性催化剂。
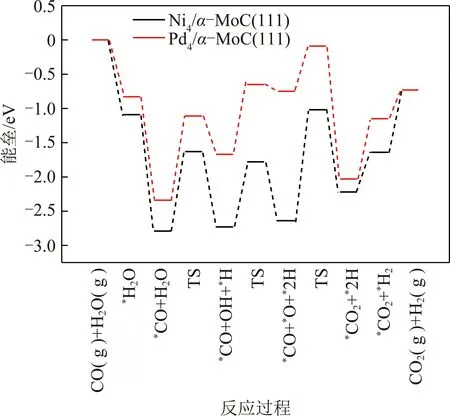
图3 WGS反应在Ni4/α-MoC(111),Pd4/α-MoC(111)上反应势能Fig.3 Potential energy of WGS on Ni4/α-MoC(111) and Pd4/α-MoC(111)
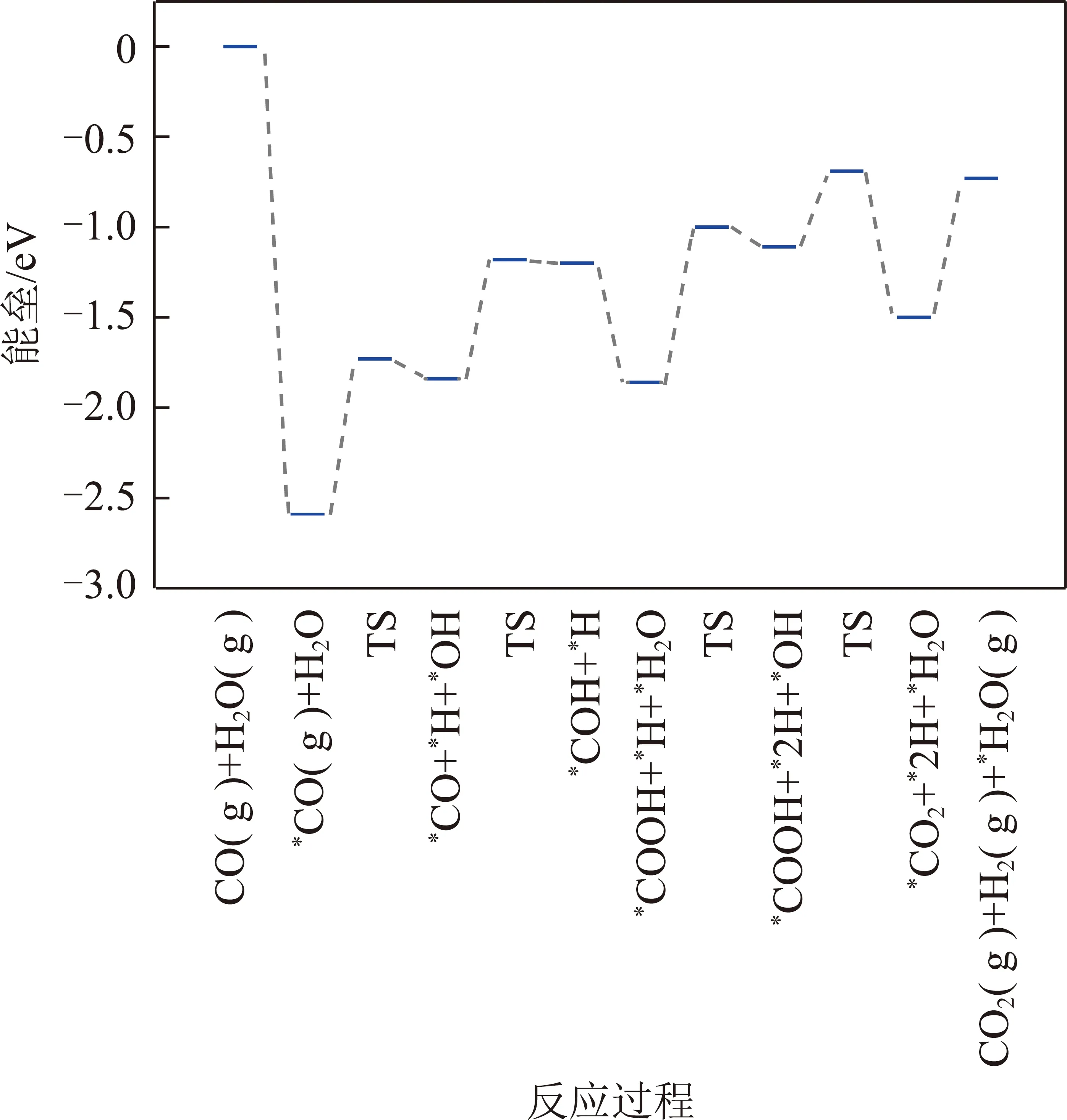
图4 WGS反应在Pt4/α-MoC(111)上反应势能Fig.4 Potential energy of WGS on Pt4/α-MoC(111)
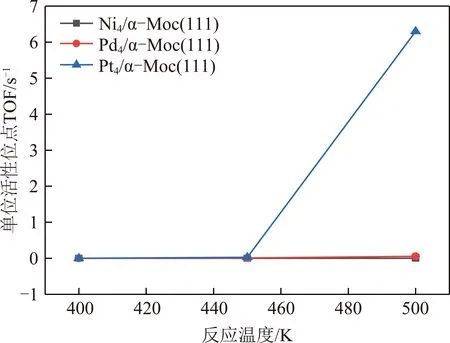
图5 WGS反应在M4/α-MoC(111)上的H2的TOFFig.5 H2 TOF for WGS reaction on M4/α-MoC(111)
3 结 论
(1)通过密度泛函理论和动力学蒙特卡洛方法探究了过渡金属Ni,Pd,Pt负载α-MoC催化剂的WGS反应的反应机理和活性。
(2)DFT计算结果发现,在Ni4/α-MoC(111)和Pd4/α-MoC(111)上,WGS反应的反应路径为氧化还原路径为CO(g)+H2O(g)→*CO+*H2O→*CO+*OH+*H→*CO+*O+*2H→*CO2+*H2→CO2(g)+H2(g);在Pt4/α-MoC(111)催化剂上,WGS反应通过羧酸盐路径发生,即CO(g)+2H2O(g)→*CO+2*H2O→*CO+2*OH+2*H→*COOH+*OH+2*H→*CO2+*H2O+*H2→CO2(g)+H2O(g)+H2(g)。
(3)动力学模拟结果表明,Pt4/α-MoC(111)具有较高的催化活性。标准大气压下,反应温度在400~500 K,H2O与CO的物质的量之比为1时,H2的TOF排序:Pt4/α-MoC(111)≫Pd4/α-MoC(111)>Ni4/α-MoC(111),Pt/α-MoC是WGS反应的高活性催化剂。
