2019年广州市甲型H1N1流感病毒全基因组序列的遗传特征分析
2021-05-06陈艺韵陆剑云张周斌李魁彪
曹 蓝,吴 迪,陈艺韵,曾 庆,夏 丹,陆剑云,狄 飚,张周斌,李魁彪
长期以来,季节性流感威胁着人类健康,每年造成严重的人群发病和死亡。甲型H1N1流感病毒自2009年首次出现以来,由于不断快速的发生基因变异,已代替了原季节性H1N1流感而持续流行。Mi L等[1]在对1977年以来全球H1N1流感病毒抗原进化模型的研究中发现,在8个主要进化模式中有6个进化模式起源于亚洲,进一步发现,我国南方地区H1N1流感病毒的进化要早于北方地区,提示南方地区H1N1流感病毒在我国H1N1流感传播进化过程起到重要作用。而广州市地处华南地区,是我国重要的经济发展中心,也是包括H1N1流感在内多种传染病流行的重要地区。早期研究发现,广州市流感流行呈现双峰的流行特点,甲型H1N1流感和季节性H3N2流感交替出现[2],同时甲型H1N1流感病毒也是近期广州市流感疫情的主要病原[3]。既往的研究中重点对HA和NA基因进行了变异分析,而对甲型H1N1基因组全序列的分子进化研究相对较少,因此本研究选取7株2019年广州市甲型H1N1流感流行株进行了全基因组测序,为全面掌握近期广州市甲型H1N1流感病毒的分子流行病学特点提供研究数据。
1 材料与方法
1.1毒株来源 收集2019年广州市流感监测哨点医院流感样病例和社区流感暴发疫情病例的呼吸道标本,通过荧光定量RT-PCR方法进行甲型H1N1、甲型H3N2和乙型流感病毒核酸检测(试剂盒购于江苏硕世生物科技公司),并将流感阳性标本接种MDCK细胞系进行病毒分离,培养产物经过血凝实验-血凝抑制试验(HA-HI)对流感病毒培养物进行鉴定(血球为自制豚鼠血,血清由中国疾病预防控制中心提供)。根据分离时间的不同,本研究中随机选取7株甲型H1N1流感病毒进行全基因组测序。
1.2全基因组序列测定 参考Deng YM等[4]关于流感病毒全基因组测序的方法,并参考近期甲型H1N1流感病毒各基因全长序列,应用Oligo 6软件设计甲型H1N1流感病毒全序列各基因片段全长测序引物(引物由广州天一辉远生物公司合成),见表1。通过RT-PCR方法扩增HA、NA、PB2、PB1、PA、NP、M和NS全长序列,阳性鉴定产物送至广州天一辉远生物公司,通过ABI 3730进行病毒一代测序。

表1 甲型H1N1流感病毒各基因片段扩增引物Tab.1 Amplification primers for gene fragments of the avian influenza A(H1N1)pdm09 virus
1.3分子特征分析 通过DNA Star7.1软件拼接全序列各基因片段,同时对各基因进行同源性分析。采用MEGA 6.0软件,以基因ORF为基本单元,对不同进化分支变异氨基酸进行分析。
1.4遗传进化分析 以GISAID数据库中历年WHO推荐的甲型H1N1流感病毒疫苗株各基因片段作为参比序列(A/Guangdong-Maonan/1536/2019 (H1N1)、A/Hawaii/70/2019(H1N1)、A/ Brisbane/02/2018 (H1N1)、A/Michigan/45/2015 (H1N1)、A/California/7/2009 (H1N1)、A /Christchurch/16/2010 (H1N1)、A/Brisbane/59/2007 (H1N1)、A/Solomon/Islands/3/2006 (H1N1)、A/New/Caledonia/20/1999 (H1N1)等)。使用MEGA 软件绘制各基因遗传进化树,参数设置为:Neighbor-joining法(参数设置为1 000 replications)及Maximum composite likelihood model核苷酸替代模型。
2 结 果
2.1总体情况 2019年累计从10 541份监测标本中检测到甲型H1N1流感阳性标本703份,其中分离甲型H1N1流感毒株70株(均感染MDCK细胞经一代培养)。本研究中7株病毒中,5株分离自流感监测病例,2株分离自流感暴发疫情病例。2株分离于2019年1月,2株分离于2019年3月,2株分离于2019年6月,1株分离于2019年12月。
2.2全基因组序列测定 全序列各基因扩增产物经毛细管电泳鉴定,经过序列拼接,获得全基因各片段开放阅读框序列(HA基因:1 701 bp、NA基因:1 410 bp、PB2基因:2 280 bp、PB1基因:2 274 bp、PA基因:2 151 bp、NP基因:1 497 bp、M1基因:759 bp、NS1基因:693 bp)。7株病毒全基因序列中,核苷酸同源性最高为PB2基因(98.8%~99.9%),最低为NA基因(97.3%~99.2%),氨基酸同源性最高为PB2蛋白(99.3%~100%),最低为NA蛋白(96.4%~99.4%),全基因组同源性见表2。
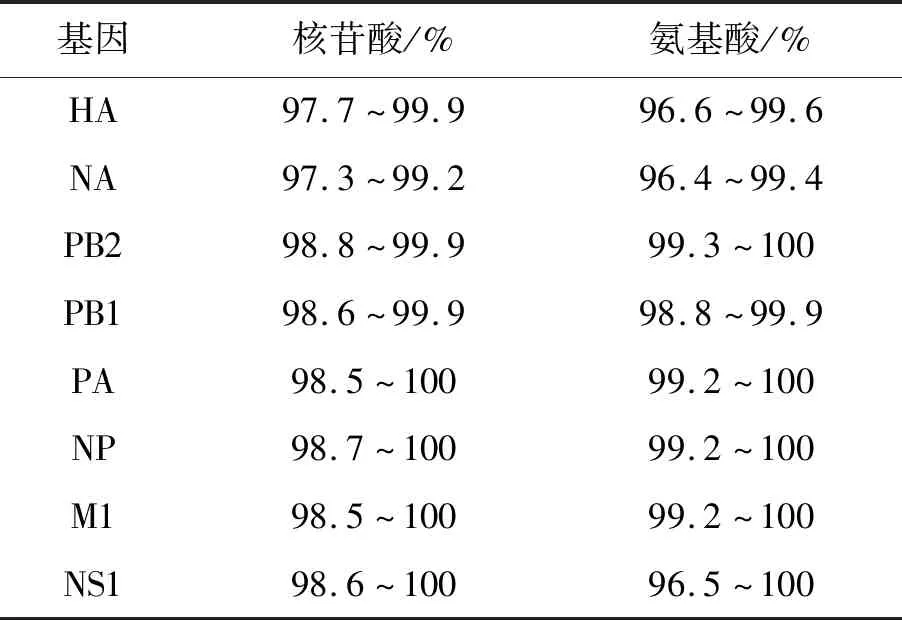
表2 2019年广州市H1N1流感分离株全序列基因的同源性分析Tab.2 Sequence similarity analysis of whole genes from novel influenza A (H1N1)pdm09 isolates in Guangzhou in 2019
2.3遗传进化分析 总体上,不同月份的H1N1流感分离株按时间分布,分别于A/Michigan/45/2015 (H1N1)、A/Brisbane/02/2018 (H1N1)、A/Guangdong-Maonan/1536/2019 (H1N1)、A/Hawaii/70/2019(H1N1)疫苗株聚为一簇,特别是2019年6月份以后的H1N1分离株与世界卫生组织2020-2021年推荐使用的疫苗推荐株A/Guangdong-Maonan/1536/2019 (H1N1)亲缘关系最近。分离于哨点医院流感样症状监测病例的毒株与社区流感暴发疫情的毒株归属于同一进化分支。全基因组遗传进化树显示,未发现不同基因来源的基因重配现象。
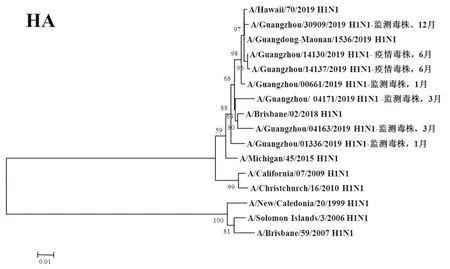
图1 2019年广州市甲型H1N1流感分离株HA基因遗传进化树Fig.1 Genetic evolution analysis of HA genes from novel influenza A (H1N1)pdm09 isolates in Guangzhou in 2019
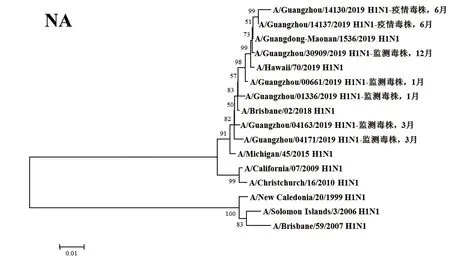
图2 2019年广州市甲型H1N1流感分离株NA基因遗传进化树Fig.2 Genetic evolution analysis of NA genes from novel influenza A (H1N1)pdm09 isolates in Guangzhou in 2019
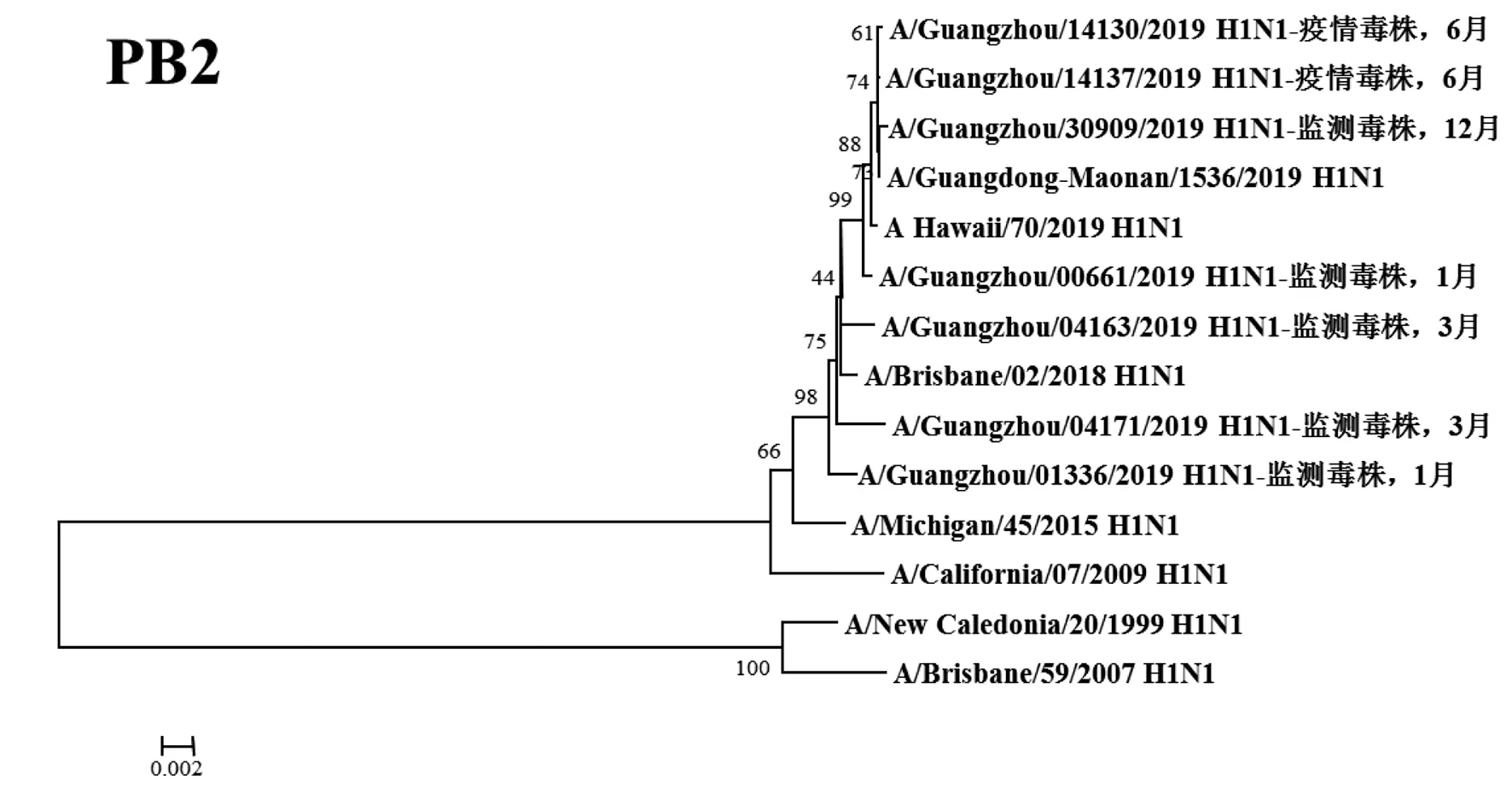
图3 2019年广州市甲型H1N1流感分离株PB2基因遗传进化树Fig.3 Genetic evolution analysis of PB2 genes from novel influenza A (H1N1)pdm09 isolates in Guangzhou in 2019

图4 2019年广州市甲型H1N1流感分离株PB1基因遗传进化树Fig.4 Genetic evolution analysis of PB1 genes from novel influenza A (H1N1)pdm09 isolates in Guangzhou in 2019
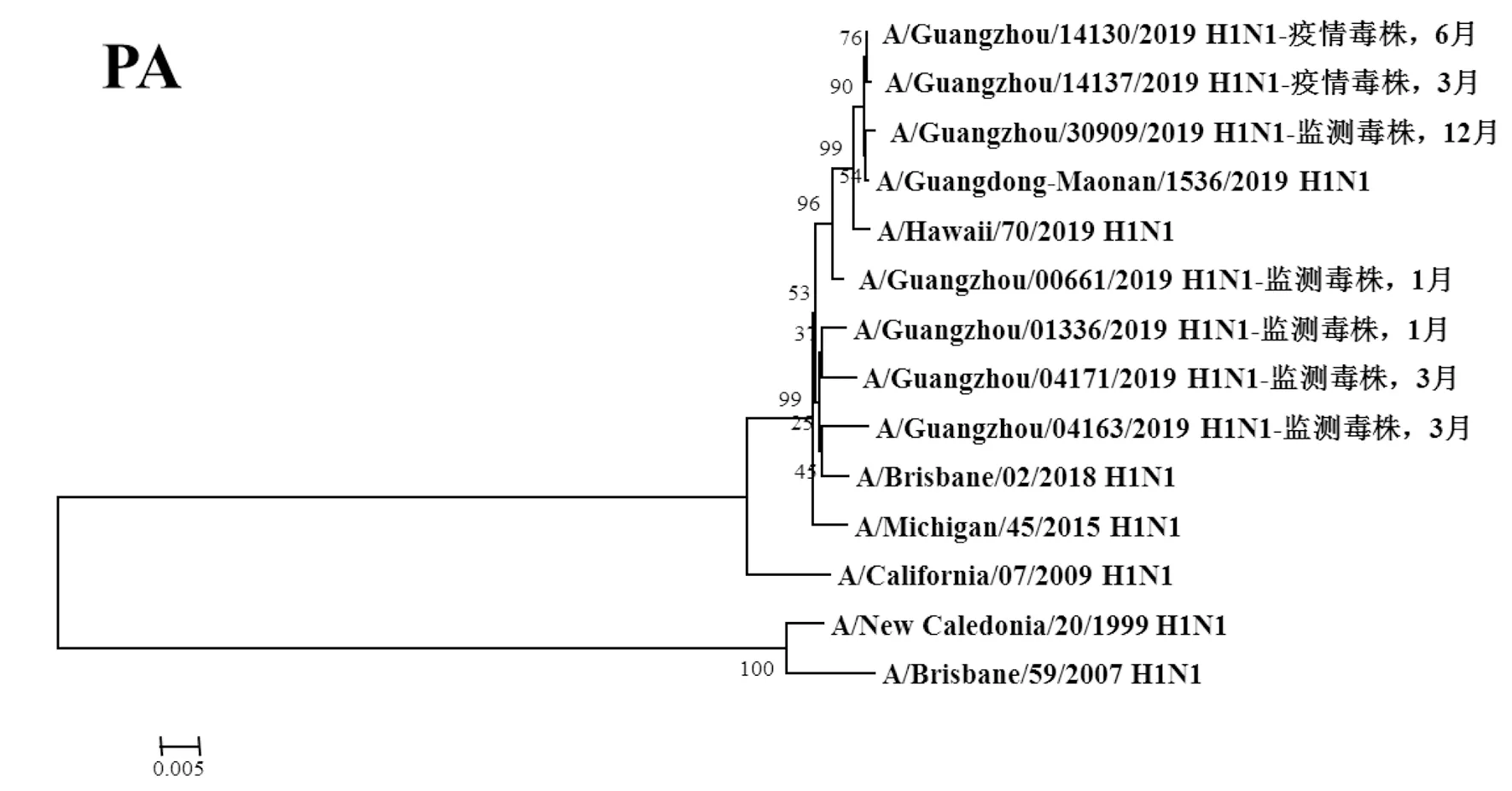
图5 2019年广州市甲型H1N1流感分离株PA基因遗传进化树Fig.5 Genetic evolution analysis of PA genes from novel influenza A (H1N1)pdm09 isolates in Guangzhou in 2019
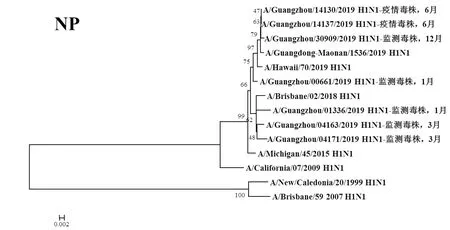
图6 2019年广州市甲型H1N1流感分离株NP基因遗传进化树Fig.6 Genetic evolution analysis of NP genes from novel influenza A (H1N1)pdm09 isolates in Guangzhou in 2019
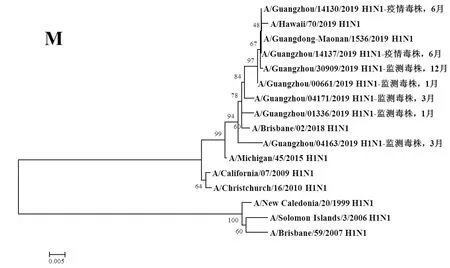
图7 2019年广州市甲型H1N1流感分离株M基因遗传进化树Fig.7 Genetic evolution analysis of M genes from novel influenza A (H1N1)pdm09 isolates in Guangzhou in 2019
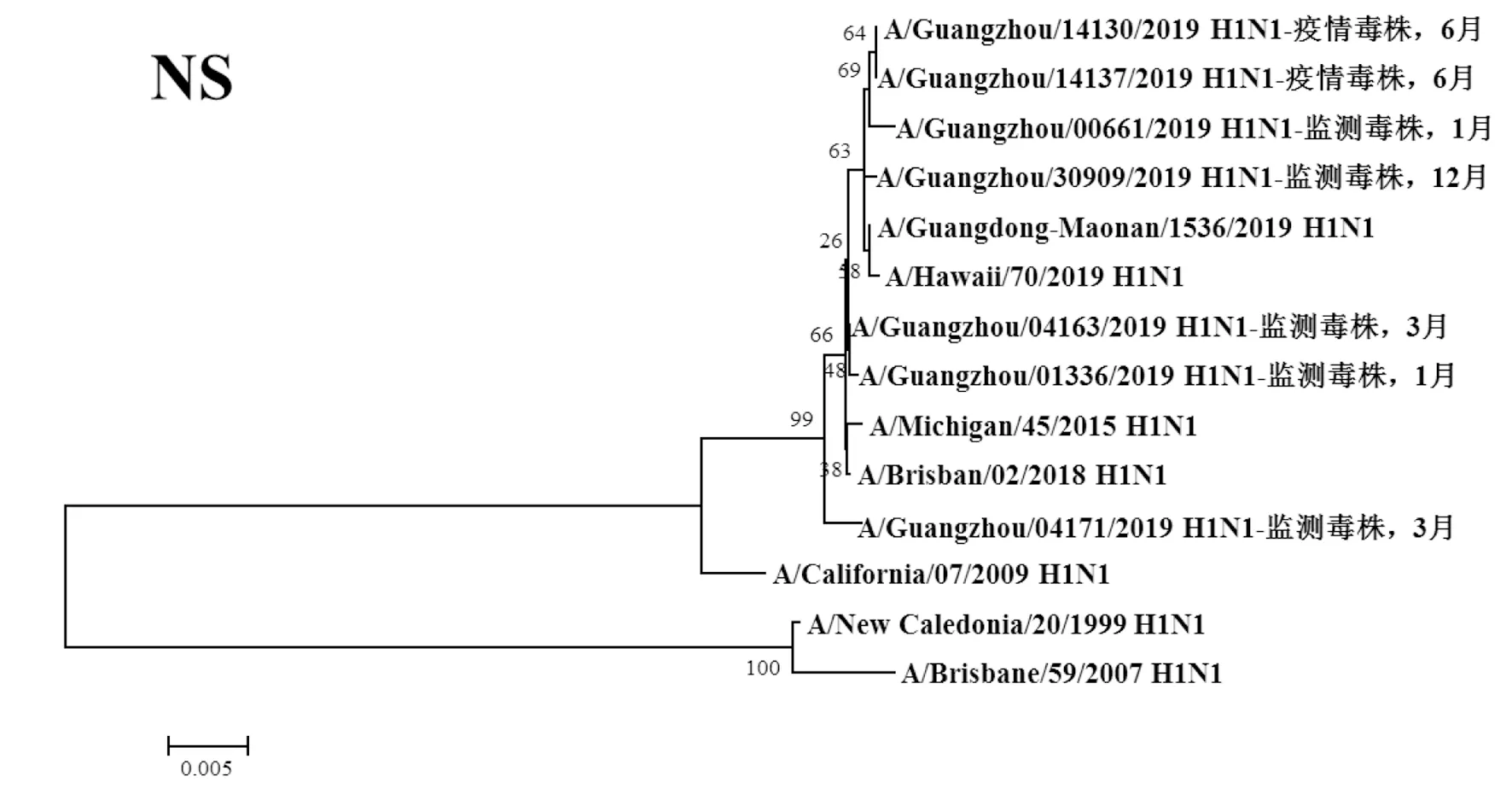
图8 2019年广州市甲型H1N1流感分离株NS基因遗传进化树Fig.8 Genetic evolution analysis of NS genes from novel influenza A (H1N1)pdm09 isolates in Guangzhou in 2019
2.4全基因分子特征分析 HA蛋白抗原位点分析结果显示,与2020-2021年疫苗推荐株A/Guangdong-Maonan/1536/2019 (H1N1)相比,01336、04163、04171分离株发生I202T变异,14130、14137、30390分离株发生D204A变异。大部分毒株HA蛋白有7个糖基化位点,04171分离株发生104-NGT糖基化位点缺失。在NA蛋白神经氨酸酶抑制剂耐药位点上,所有分离株未发生耐药突变。
根据全基因进化树结果显示,A/Guangzhou/00661/2019 (H1N1)、A/Guangzhou/14130/2019 (H1N1)、A/Guangzhou/14137/2019 (H1N1)和A/Guangzhou/30309/2019 (H1N1)毒株全基因片段均与我国2020-2021年疫苗推荐株A/Guangdong-Maonan/1536/2019 (H1N1)归属同一进化分支,对上述4株分离株毒株全基因氨基酸位点与另3株分离株进行变异分析,结果显示,除PA、M1、M2和NS2蛋白外,其余蛋白均有不同数量的相同特异性氨基酸突变,见表3。同时对两株流感疫情分离株A/Guangzhou/14130/2019 (H1N1)和 A/Guangzhou/14137/2019 (H1N1)进行全基因氨基酸变异位点特异性分析显示,两毒株具有HA-K188E、NA-I40T、PB1-M646L、NS1-I145V等共同的氨基酸变异位点。
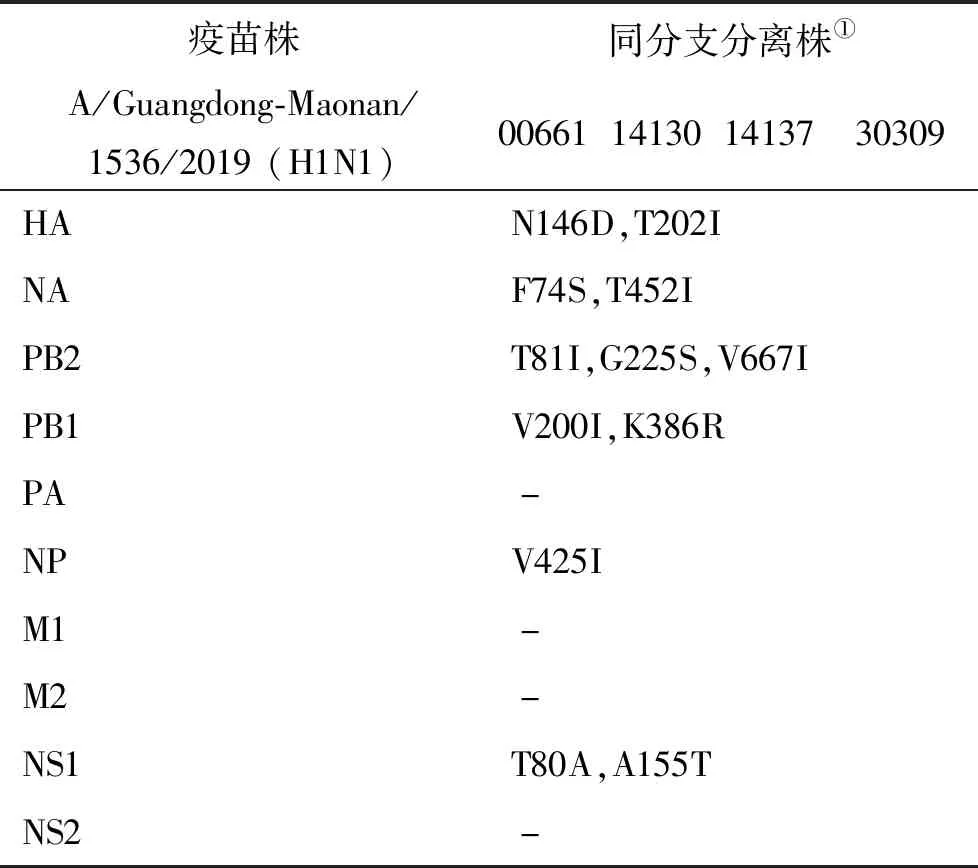
表3 同分支分离株基因组特异性氨基酸变异分析Tab.3 Analysis of genome specific amino acid variations in homocladistic isolates
3 讨 论
自2017年开始,WHO推荐北半球使用的3株H1N1疫苗株(A/Michigan/45/2015 (H1N1)(2017-2019)、A/ Brisbane/02/2018 (H1N1)(2019-2020)、A/Guangdong-Maonan/1536/2019 (H1N1)(2020-2021))亲缘关系较近,遗传进化上不断进化,2020-2021年疫苗株与2019年6月之后的流行株在全基因组上位于同一遗传进化簇上,提示2020年下半年启动使用的疫苗株与流行株匹配性较好。但该分支中00661毒株分离于2019年1月,说明广州地区甲型H1N1流感病毒流行的复杂性,不排除较早期流行株与近期流行株共同流行的情况,进而存在病毒重配风险,因此及时监测流行的变异情况、评估疫苗株与流行株的匹配性尤为重要。
广州流感流行表现为一年两次的流行高峰,本研究发现6月份以后的流行株全基因组序列在遗传进化上与疫苗株(2020-2021)共同形成独立进化分支,进一步氨基酸位点分析也显示,该分支分离株与疫苗株具有多个位点相同的特异性氨基酸突变,揭示2019年下半年流行株与疫苗株匹配性较好的分子基础。本研究中2株流感疫情流行株与门诊监测流行株高度同源,进化起源相同。但对2株疫情分离株全基因组氨基酸变异分析显示,在HA、NA、PB1和NS1蛋白存在不同于监测毒株的特异性氨基酸变异,这些位点是否与病毒传播能力相关,进而引发流感疫情暴发,需要进一步研究。
由于流感病毒基因组分节段的特点,当不同亚型流感病毒共同感染宿主细胞时可发生基因重配[5],这也是流感病毒不断进化的重要方式之一,新重配的病毒常引起人间流感的大流行,在历史上有3次流感大流行就是由重配流感病毒所引起[6-7]。2009年引起全球广泛流行的pdm2009 H1N1(本研究中也称“甲型H1N1”)即是由南美H1N1禽流感病毒的PB2、PA基因,人H3N2流感病毒的PB1基因、南美H1N1经典猪流感病毒的HA、NP、NS和欧亚类禽H1N1猪流感病毒的NA、M基因重配进化而来[8]。大量研究发现,pdm2009 H1N1流感病毒宿主范围多样,可以感染火鸡[9]、犬[10]、猫[11-13]、野鸟[14]等,提示pdm2009 H1N1流感病毒有重配产生新型流感病毒的可能[15]。因此开展甲型H1N1流感病毒全基因组进化分析对新型重配病毒的发现有着重要意义。在本研究中所选取的9株2019年广州市甲型H1N1流感病毒未发现不同基因来源的基因重配现象。2020年新型冠状病毒在全球广泛流行,我国及时启动了全面且有效的防疫对策,在采取高风险地区封闭管理、学校和托幼机构休学、加强佩戴口罩和卫生消毒等一系列防疫措施后,我国的新冠病毒传播得以有效控制,同时也控制了包括甲型H1N1流感病毒在内的呼吸道病原体的流行和传播。2020年广州市流感监测数据显示,截止目前尚未监测到甲型H1N1流感病毒,但随着复工复产、人流活动的增加,下一阶段是否会发生甲型H1N1流感病毒流行,以及继续2019年下半流行株持续流行,还是在此基础上出现新变异株,需要持续监测。
利益冲突:无
