布鲁氏菌微滴式数字PCR方法的建立与应用
2021-05-06田国忠薛盼盼朴东日赵鸿雁杨晓雯张京云
田国忠,姜 海,薛盼盼,朴东日,赵鸿雁,杨晓雯,张京云
布鲁氏菌病(Brucellosis)简称布病,是由细胞内寄生的布鲁氏菌(Brucella)引起的一种人兽共患传染-变态反应性疾病,可感染多种家畜、家禽和野生动物,其中与人类密切相关的动物主要有羊、牛、猪和犬等。病畜的血液、分泌物、排泄物、流产物及乳类含有的致病菌,可在动物间传播,造成感染发病;传染到人,可引起人体感染,出现临床症状。目前布鲁氏菌的检测方法主要有:细菌的分离培养、布鲁氏菌抗体检测(试管凝集试验)和分子生物学检测(常规PCR、荧光定量PCR和环介导等温扩增技术)等。细菌的分离培养和血清学诊断技术是经典方法,但存在着检测周期长、操作繁琐、易漏检和潜在生物危害等缺点。常规PCR方法需要凝胶电泳分析,容易发生PCR产物的实验室污染,并且灵敏性低;荧光定量PCR具有较高的灵敏性,但是血液等标本中经常存在的PCR抑制因素影响了荧光定量PCR的实际应用[1-2];环介导等温扩增技术由于引物间的非特异性扩增而易产生假阳性结果。微滴式数字PCR方法(Droplet digital PCR,ddPCR)是近年来发展起来的快速、准确、可实现DNA绝对定量的PCR方法,已经广泛应用于细菌和病毒的检测[3]。本研究建立了布鲁氏菌的ddPCR方法,可实现对布鲁氏菌核酸DNA快速、准确的定量检测,适合布病临床标本和布病的分子流行病学调查。
1 材料与方法
1.1仪器与试剂 微滴式数字PCR系统QX200 Droplet Digital PCR(Bio-Rad,美国)包括微滴生成仪、微滴分析仪、封膜仪和Quanta Soft V1.7.4软件。其它设备:荧光定量PCR仪(LightCycler 4800II,Roche,美国),普通PCR扩增仪(LabcyclerK,SENAQUEST,德国)和DNA微量分光光度测定仪(spectrophotometer NanoDrop 1000,美国)。荧光定量PCR试剂Premix Ex TaqTM(Probe qPCR)(宝生物工程(大连)有限公司,中国);ddPCR试剂和耗材,包括ddPCRTMSupermix for Probes、微滴生成油、微滴生成卡槽、微滴生成卡槽胶垫和锡纸膜,均由美国Bio-Rad公司生产;细菌基因组DNA提取试剂盒和血液基因组DNA提取试剂盒购自天根生化科技(北京)有限公司。
1.2引物和探针 荧光定量PCR和ddPCR使用相同的引物和探针[4],由生工(上海)生物科技有限公司合成。其序列为:上游引物B4 5′-GCTCGGTTGCCAATATCAATGC-3′,下游引物B5 5′-GGGTAAAGCGTCGCCAGAAG-3′,探针probe 5′FAM-AAATCTTCCACCTTGCCCTTGCCATCA-3′ BHQ1。
1.3菌株和标本 标准菌株S3(猪种布鲁氏菌生物3型)用于ddPCR反应体系的优化和灵敏性评价。特异性评价使用了7株其它种属的菌株,包括钩端螺旋体、小肠结肠炎耶尔森菌(O∶9)、霍乱弧菌(非O1/非O139血清型)、大肠埃希菌(O∶157)、结核分枝杆菌、炭疽芽孢杆菌和伤寒沙门菌各一株,其菌株核酸由中国疾病预防与控制中心传染病预防控制所相关科室提供。采集于2014年3月一起羊种布鲁氏菌病暴发疫情的17份工作人员血液标本核酸和5份羊血液标本核酸DNA用于ddPCR方法的实际检测效果评价。该疫情的相关流行病学调查资料和样本DNA的提取方法详见文献[5]。
1.4灵敏性评价 将经过连续3代培养的标准菌株S3的新鲜培养物,用0.85%的生理盐水制成菌悬液,取200 μL菌悬液,10 000 r/min离心1 min,弃去上清,沉淀物按照细菌基因组DNA提取试剂盒说明书进行DNA提取,洗脱体积为200 μL;应用DNA微量分光光度测定仪测定DNA浓度,并对DNA进行2倍的系列稀释。对各个稀释度的DNA分别进行ddPCR和荧光定量PCR检测,每个稀释度进行4次平行实验,取4次检测的平均值分析线性区间和灵敏性。
1.5特异性评价 将钩端螺旋体、小肠结肠炎耶尔森菌(O∶9)、霍乱弧菌(非O1/非O139血清型)、大肠埃希菌(O∶157)、结核分枝杆菌、炭疽芽孢杆菌和伤寒沙门菌的核酸DNA,分别稀释到1 ng/μL、10 pg/μL和10 fg/μL 3个浓度,每个稀释度进行4次重复检测,以评价荧光定量PCR和ddPCR的特异性。
1.6荧光定量PCR反应条件 20 μL的反应体系包含:Premix Ex TaqTM(Probe qPCR) (2×) l0 μL,上、下游引物(10 mol/L)各0.8 μL ,探针(10 mol/L)0.4 μL,DNA模板量为1 μL,补充无菌去离子蒸馏水至20 μL体积。反应条件:95 ℃ 3 min;95 ℃ 5 s,60 ℃ 20 s,40个循环,在退火阶段检测荧光信号。
1.7ddPCR反应条件 20 μL反应体系包括:ddPCRTM预混液(Supermix for Probes)10 μL;核酸DNA模板1 μL;上、下游引物和探针(待优化);补充无菌去离子蒸馏水至20 μL体积。随后将20 μL反应液和70 μL微滴生成油加入到微滴生成卡槽内,盖上胶垫,放入微滴生成仪中产生微滴。随后将生成的微滴用移液器转移到96孔PCR板中,盖上锡纸膜,用封膜仪封口后,放在热循环PCR仪中运行。ddPCR扩增条件为:95 ℃ 预变性10 min;95 ℃变性30 s,退火温度(待优化)1 min,循环40次;最后98 ℃酶失活10 min。ddPCR扩增完成后,将96孔PCR板放在微滴分析仪中读取荧光信号,通过QuantaSoftV1.7.4软件对结果进行分析。
2 结 果
2.1ddPCR反应体系和扩增条件的优化 在20 μL反应体系中,固定引物(10 mol/L)加入量为1.6 μL,设置探针(10 mol/L)加入量分别为0.3 μL、0.5 μL、0.8 μL、1.0 μL、1.2 μL、1.6 μL和1.8 μL。如图1A所示,各探针浓度均能明确区分阳性微滴和阴性微滴。从减少“拖尾”现象和扩增效果的角度,选择使用的探针浓度为0.4 mol/L(即加入0.8 μL探针)。在20 μL反应体系中,固定探针(10 mol/L)加入量0.8 μL,设置引物(10 mol/L)加入量分别为0.3 μL、0.5 μL、0.8 μL、1.0 μL、1.2 μL、1.6 μL和1.8 μL,结果如图1B所示。从减少“拖尾”现象和扩增效果的角度,选择使用的引物浓度为0.8 mol/L(即加入引物1.6 μL)。在20 μL反应体系中,探针(0.8 μL)和引物(1.6 μL)加入量固定不变,设置退火温度分别为55 ℃、56 ℃、57 ℃、59 ℃、60 ℃、61 ℃和62 ℃,如图1C所示,各温度均能明确区分阳性微滴和阴性微滴,从减少非特异扩增和扩增效果的角度,选用的退火温度为60 ℃。
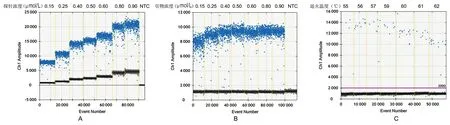
A:探针浓度优化:引物浓度固定为0.8 mol/L,各孔使用的探针浓度在图上部标出。B:引物浓度优化:探针浓度固定为0.4 mol/L,各孔使用的引物浓度在图上部标出。C:退火温度优化:引物和探针浓度分别为0.8 mol/L和0.4 mol/L,退火温度在图上部标出。图1 ddPCR引物和探针浓度以及退火温度的优化Fig.1 Optimization of the primer and probe concentrations and the annealing temperature for ddPCR
综合考虑荧光信号强度、稳定性和拖尾现象等因素,优化后的反应体系为:ddPCRTM Supermix for Probes l0 μL,上、下游引物(10 mol/L)各1.6 μL,探针(10 mol/L)0.8 μL,核酸DNA模板1 μL补充无菌去离子蒸馏水至20 μL体积。优化后的扩增条件为:95 ℃预变性10 min;95 ℃变性30 s,60 ℃退火1 min,循环40次;98 ℃酶失活10 min,4 ℃保存至下一步操作。在上述条件下,荧光信号集中,阳性微滴和阴性微滴明显分开,无拖尾现象,形成最佳的微滴数和扩增效果。
2.2线性区间和灵敏性 标准菌株S3的基因组DNA浓度为30.85 ng/μL,将DNA 2倍梯度稀释,进行ddPCR线性区间和灵敏性的评价(图2)。当加入ddPCR反应体系的核酸DNA在0.96 ng/反应~3.68 fg/反应时,ddPCR测得值为2.00×105拷贝/反应~1拷贝/反应,ddPCR测得值的对数值(y)与DNA浓度的对数值(x)有较好的线性关系(y=0.998 6x-0.612 9,R2=0.998 2)。加入每个反应的模板DNA量高于0.96 ng时,因超过了ddPCR的测量能力范围,ddPCR不能进行定量,而是给出2.00×107拷贝/反应的常数值。
标准菌株S3的基因组DNA2倍梯度稀释液平行进行荧光定量PCR检测(图2)。当加入反应体系的核酸DNA为30.85 ng/反应~3.68 fg/反应时,荧光定量PCR检测均有扩增曲线,对应的Ct值为11.50~36.20,Ct值(y)与DNA浓度的对数值(x)有较好的线性关系(y=-3.620 8x+38.343,R2=0.998 8),线性区间比ddPCR宽。
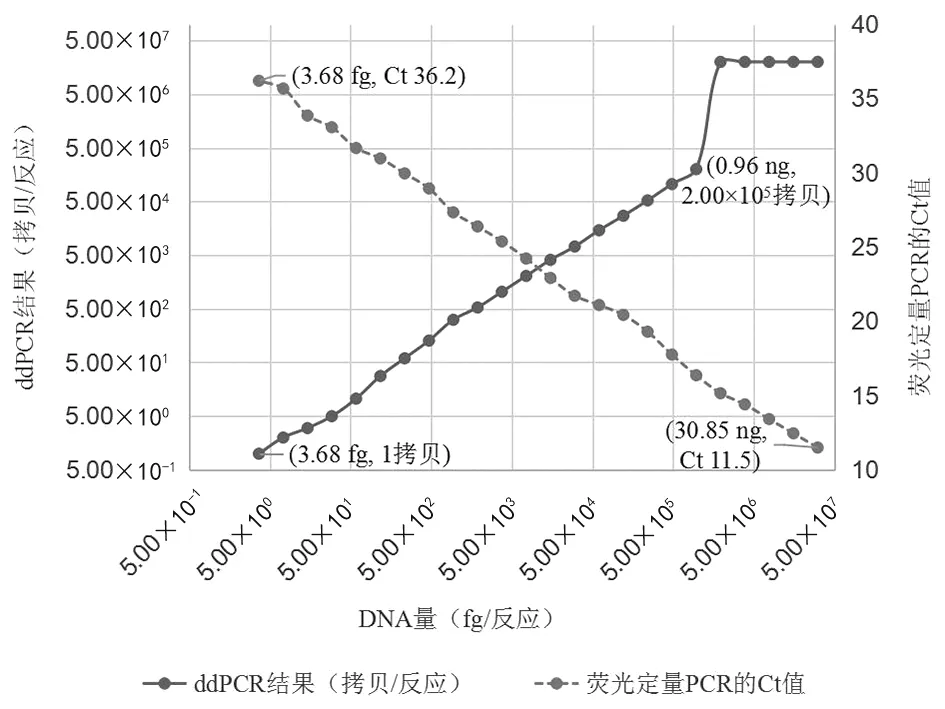
图2 ddPCR和荧光定量PCR检测结果随布鲁氏菌DNA量的变化Fig.2 Changes in detection results of ddPCR and real-time PCR with the amount of Brucella DNA
布鲁氏菌核酸DNA浓度、ddPCR结果和荧光定量PCR结果见表1,3部分数据反映的变化趋势基本一致。ddPCR可以直接给出靶标基因拷贝数,实现绝对定量;而荧光定量PCR需要建立标准曲线并根据公式进行换算才能获得靶标基因拷贝数。
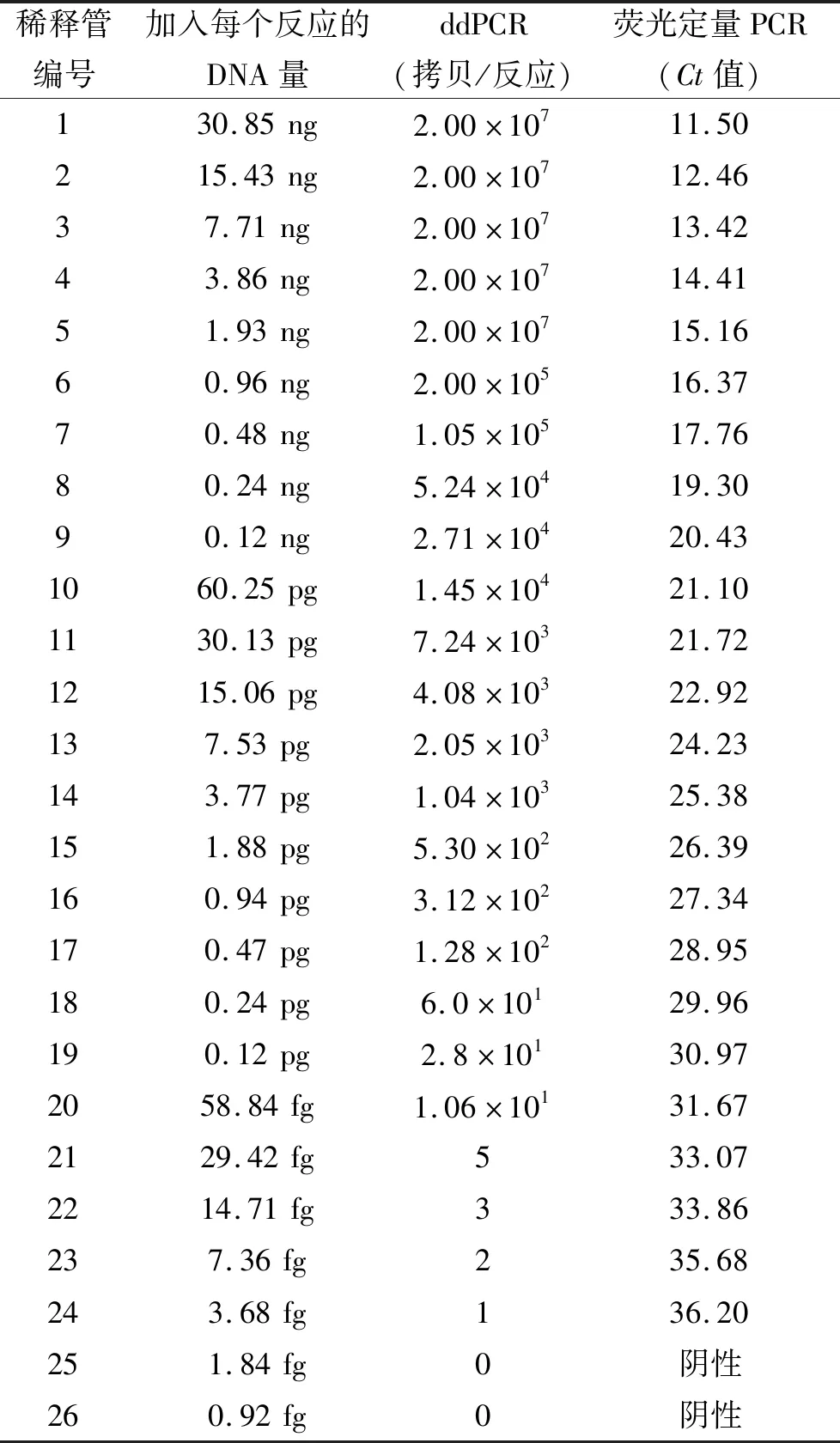
表1 ddPCR和荧光定量PCR检测结果随布鲁氏菌DNA量的变化Tab.1 Changes in detection results of ddPCR and real-time PCR with the amount of Brucella DNA
2.3特异性 以来自非布鲁氏菌种属的7株菌进行特异性评价。这些菌株3个浓度(1 ng/μL、10 pg/μL和10 fg/μL)的核酸DNA分别以荧光定量PCR和ddPCR进行4次重复检测,荧光定量PCR皆无扩增曲线,ddPCR皆未检测到阳性微滴,提示所使用的引物和探针具有高度特异性。
2.4疫情相关血液标本DNA的检测 2014年发生的布病疫情中,18名工作人员的血液标本中有2个标本、8只羊的血液标本中有3个标本培养出细菌,均为羊种布鲁氏菌生物3型菌株[5]。血液标本提取的核酸DNA于-40 ℃冻存,因部分核酸DNA缺失,本研究仅采用17名工作人员和5只羊的血液标本核酸。标本核酸于4 ℃融化后以ddPCR方法进行检测,发现16份人血液标本为阳性,DNA模板浓度为5~65拷贝/mL血液,平均29拷贝/mL血液,只有1份人血液标本在ddPCR检测中为阴性,此标本在培养法和试管凝集检测中皆为阴性;5份羊血液标本全部为阳性,DNA模板浓度为25~245拷贝/mL血液,平均98拷贝/mL血液。
3 讨 论
数字PCR从概念提出到商业化生产在短短几十年间经历了飞速发展。目前数字PCR主要分为3种:微孔数字PCR、微滴式数字PCR和微腔数字PCR。其中,ddPCR是目前相对成熟的数字PCR技术,其原理是利用油包水乳化,将一定的反应体系分散成上万个微滴,每个微滴形成一个独立的、包含或不包含模板DNA的反应体系;PCR扩增后,读取阳性微滴数,根据泊松分布计算出样本中的DNA分子数[6-7]。ddPCR已经被用于多种传染性疾病病原体的检测,例如HIV-1[8]、结核分枝杆菌[8]等。随着技术的进一步成熟,ddPCR将在传染病检测领域发挥更重要的作用。
布鲁氏菌的bcsp31基因在布鲁氏菌属中具有高度保守性和种属特异性,最常用于人布鲁氏菌的诊断[10];本研究使用针对bcsp31基因的引物和探针,探索最优反应体系和条件,建立了探针法的ddPCR方法。以纯培养的标准菌株核酸DNA进行评价,本研究建立的ddPCR方法灵敏性高,可以实现单拷贝模板的检测。在模板量为0.96 ng/反应~3.68 fg/反应时,ddPCR测得值与模板DNA量有较好的对数线性关系。平行进行的荧光定量PCR检测也达到了3.68 fg/反应的灵敏性,且线性范围更宽(30.85 ng/反应~3.68 fg/反应)。但是,荧光定量PCR对模板的定量需要利用已知浓度的模板建立标准曲线,并获得待检测标本的信号阈值。信号阈值和标准曲线是测试和仪器专用的,不同的检测平台、探针类型和校准标准都会影响所获得的信号阈值,从而影响原始标本中模板的定量。另外,当模板浓度接近定量下限时,变异系数通常很大。模板中含有扩增抑制因素时,荧光定量PCR的信号阈值会增加,导致对模板量的估计低于实际值。ddPCR的定量是通过分析扩增终点时的产物,而非分析实时扩增过程。扩增抑制因素的存在可能使阳性微滴的荧光信号值降低,但是对阳性微滴的数量影响较小,因此对定量结果的影响较小;同理,ddPCR定量结果受扩增效率的影响也较小[11]。在特异性方面,选择来自7个其它种属的菌株进行测试,荧光定量PCR和ddPCR检测结果皆为阴性,说明使用的引物和探针具有较好的特异性。
虽然ddPCR具有潜在的应用价值,但与荧光定量PCR相比,该方法目前存在一些缺点:需要昂贵的专用设备、试剂和耗材的价格较高、对操作者的技术要求高、检测通量低,这些因素都限制了该技术的推广应用。结果报告方面,如何合理地审核和报告低丰度的检测结果,并做好质量控制,是该方法应用于临床实践前需要考虑的要点。
利益冲突:无
