多倍体同源区段二代测序生物信息学分析关键参数优化
2021-04-16武建楠陈肯王欢庞铂实周宇荀肖君华李凯
武建楠,陈肯,王欢,庞铂实,周宇荀,肖君华,李凯*
(1.东华大学化学化工与生物工程学院,上海 201620;2.上海农林职业技术学院,上海 201699)
单核苷酸多态性(Single nucleotide polymorphism,SNP) 是由单碱基突变引起的DNA 序列多态性, 作为新一代遗传标记具有数量丰富、分布广泛和覆盖度高等特点[1]。 与其他分子标记相比,SNP 标记在基因分型过程可以做到高度自动化、高通量检测[2],已成为包括玉米在内的二倍体植物研究中的首选标记类型[3]。 2010 年滑峰等[4]发现CYP2A12基因的同源序列对SNP 分析产生严重影响。异源多倍体SNP 开发与鉴定的困境主要在于靶区段常存在大量同源序列, 存在SNP、部分同源序列变异(Homoeologous sequence variant,HSV)[5]和旁系同源序列变异(Paralogous sequence variant,PSV)[6]等多种变异类型。 在多倍体遗传研究中,80%以上假阳性SNP 是HSV 和PSV 而不能用于开发遗传标记[7]。
多倍体物种SNP 分型的常用技术包括Sanger 测序、SNP 芯片、 竞争性等位基因特异性PCR(Kompetitive allele-specific polymerase chain reaction,KASP)等。然而,它们依赖特异性扩增或特异性杂交, 而多倍体物种亚基因组间高度同源, 靶向特异性大大降低。 第二代测序(Next generation sequencing,NGS) 技术的发展使得测序费用大幅度降低,测序效率极大提高。 目前已有研究者尝试采用生物信息学方法区分多倍体SNP 和HSV(也称部分同源SNP),即通过不同的过滤方法[8]识别并消除部分同源SNP。 该方法在不同倍性的物种如花生(异源四倍体)、小麦(异源六倍体)和草莓(异源八倍体)的模拟数据中均表现出很高的准确性[9],有望得到广泛应用。
本研究的对象为异源四倍体陆地棉(Gossypium hirsutumL.),是由亚洲棉(G. arboreum,AA)和雷蒙德氏棉(G. raimondii,DD)杂交加倍而来[10]。在A12 染色体上随机选取了3 个靶标区段,并对136 个陆地棉样本的DNA 用多重PCR 靶向扩增子测序[11]。 将参考基因组作为关键参数进行调整优化,对其分型结果进行比较,研究同源区段对多倍体SNP 鉴定与生物信息学分析的影响。 优化生物信息学方案后获得了准确的分型信息,提高了SNP 分型的准确度,为棉花等多倍体作物全基因组SNP 基因型分析以及定制育种打包检测奠定了基础。
1 材料与方法
1.1 植物材料与DNA 提取
参考序列来源于陆地棉参考基因组Ghir-NAU (https://cottonfgd.org/about/download.html)。在136 个不同陆地棉样本中,采用匡猛等[12]的方法快速提取DNA,DNA 的OD260/OD280比值为1.8~2.0, 通过琼脂糖凝胶电泳检测DNA质量。
1.2 文库制备与测序
三个靶向扩增子区段均位于陆地棉A12 染色体(表1)。 利用本实验室前期设计的靶向测序的多重PCR 引物系统TASPrimer[13],对潜在变异位点设计特异性PCR 引物(表2),由上海百力格生物技术有限公司合成引物。PCR 反应的产物大小为215~237 bp,引物长度为19~30 bp,退火温度为57~63 ℃,GC 含量为28%~63%, 并设计了136 对含有索引序列和通用序列的条形码引物来区分不同样本, 使用Illumina 公司的P5、P7 建库引物和多重PCR 法[14]建库。将不同样本混合之后,使用琼脂糖凝胶对混合文库进行电泳分离,切胶回收纯化文库。 建库产物送金唯智生物(苏州金唯智生物科技有限公司)进行高通量测序(测序仪为Illumina X-10),上机前用Agilent 2100 Bioanalyzer 对该测序文库进行质量控制。
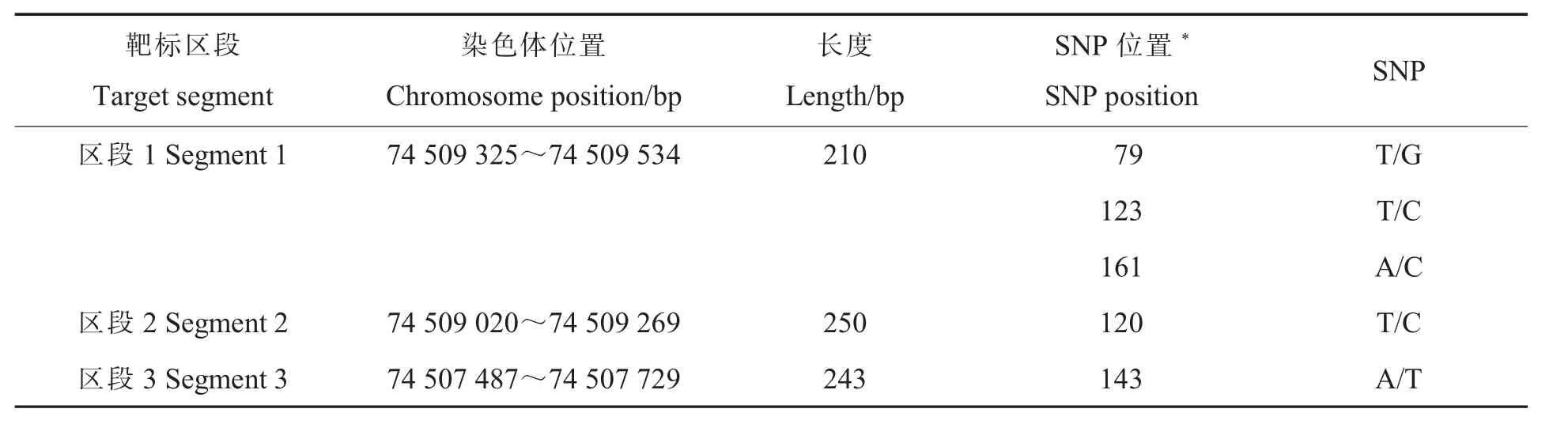
表1 靶标区段及潜在SNP 变异位点基本信息Table 1 Basic information of target segment and potential SNP variant sites
1.3 生物信息分析流程
基于已标注SNP 位点的参考基因组、原始数据和棉花基因组数据库CottonFGD[15],对关键参数即参考基因组(Reference)调整优化进行生物信息学分析, 流程如图1 所示。 原始数据含136个样本, 用FastQC 软件进行质量控制,用Cutadapt 软件切去接头序列,最终得到有效数据样本130 个。
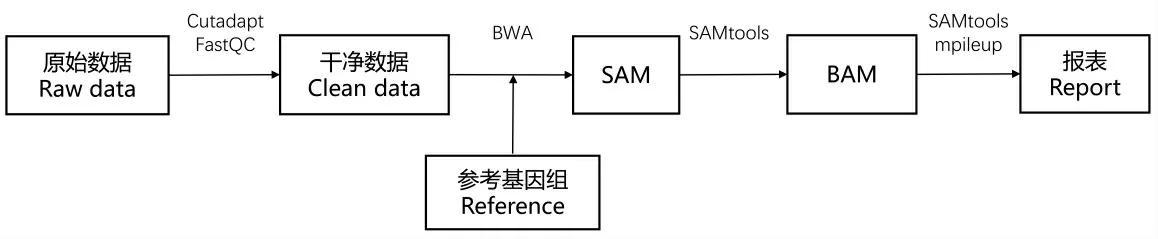
图1 生物信息分析流程Fig. 1 Bioinformatics analysis process
采用BWA 软件[16]与参考基因组进行比对得到SAM 文件,用自定义脚本对匹配的读长(Read)数进行统计生成比对数据表,并用SAMtools[17]转化为BAM 文件。 使用SAMtools mpileup 命令对目标SNP 位点进行变异分析,并分别计算比对到区段1 、区段2 和区段3 的每个样本对应读长的r值,生成散点图。r=Nv/Nt,其中Nv是变异读长数,Nt是比对到参考基因组的读长总数。r值是判断杂合性的指标[18],若用R 表示参考碱基、A 表示变异碱基,那么:当0<r≤0.2 时,表示某一样本中该位点为纯合基因型且与参考序列一致即RR;当0.8≤r<1 时,表示该位点为纯合变异基因型AA;r为其他数值时, 表示此样本该位点为杂合基因型RA。
再用GATK[19]对区段中的所有位点进行变异分析,最后生成报表检验基因型。 GATK 报表为vcf 格式,其中0/0 或1/1 均表示纯合,分别与参考序列或变异序列一致;0/1 表示杂合。
在棉花数据库(https://cottonfgd.org/)对靶标区段进行比对,确定高相似度的同源序列。 将靶标区段与同源区段分别作为参考序列进行优化调整分析。
2 结果与分析
2.1 基于靶标区段作为参考序列的分析
样本总读长数为11 031 472, 比对到3 个靶标区段的读长相对接近, 共约占总测序读长的70%,3 个扩增子覆盖率为100%。
表1 中3 个区段5 个潜在SNP 位点的有效测序深度均值分别为8 400×、8 534×、8 403×、7 716×、12 793×。 利用SAMtools 对3 个靶标区段所有目标SNP 位点进行变异分析, 结果显示:区段1 和区段2 的目标SNP 位点的r值接近1或0,说明基因型纯合;区段3 的143 位点的r值大多接近0.5,表明基因型多为杂合(图2)。
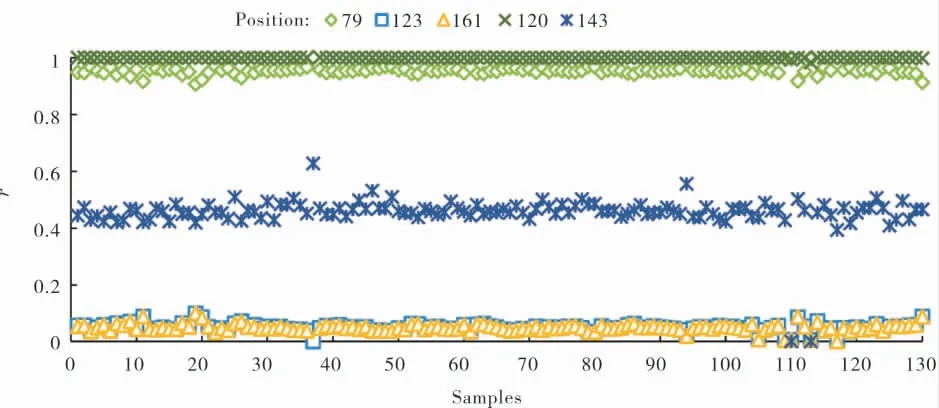
图2 3 个靶标区段目标SNP 位点等位基因的读长所占比例Fig. 2 Proportion of the target SNP locus alleles reads in the three target segments
用GATK 进行全片段SNP 检出(表3),结果表明:区段1 的3 个潜在SNP 位点和区段2 的1个潜在SNP 位点在130 个样本中100%为纯合子;而区段3 的潜在143 位点在130 个样本中有近99%为杂合,只在1 个样本中为纯合;此外,在区段3 上还检出了5 个新的变异位点(位点48、78、174、180、182)且均为杂合子。
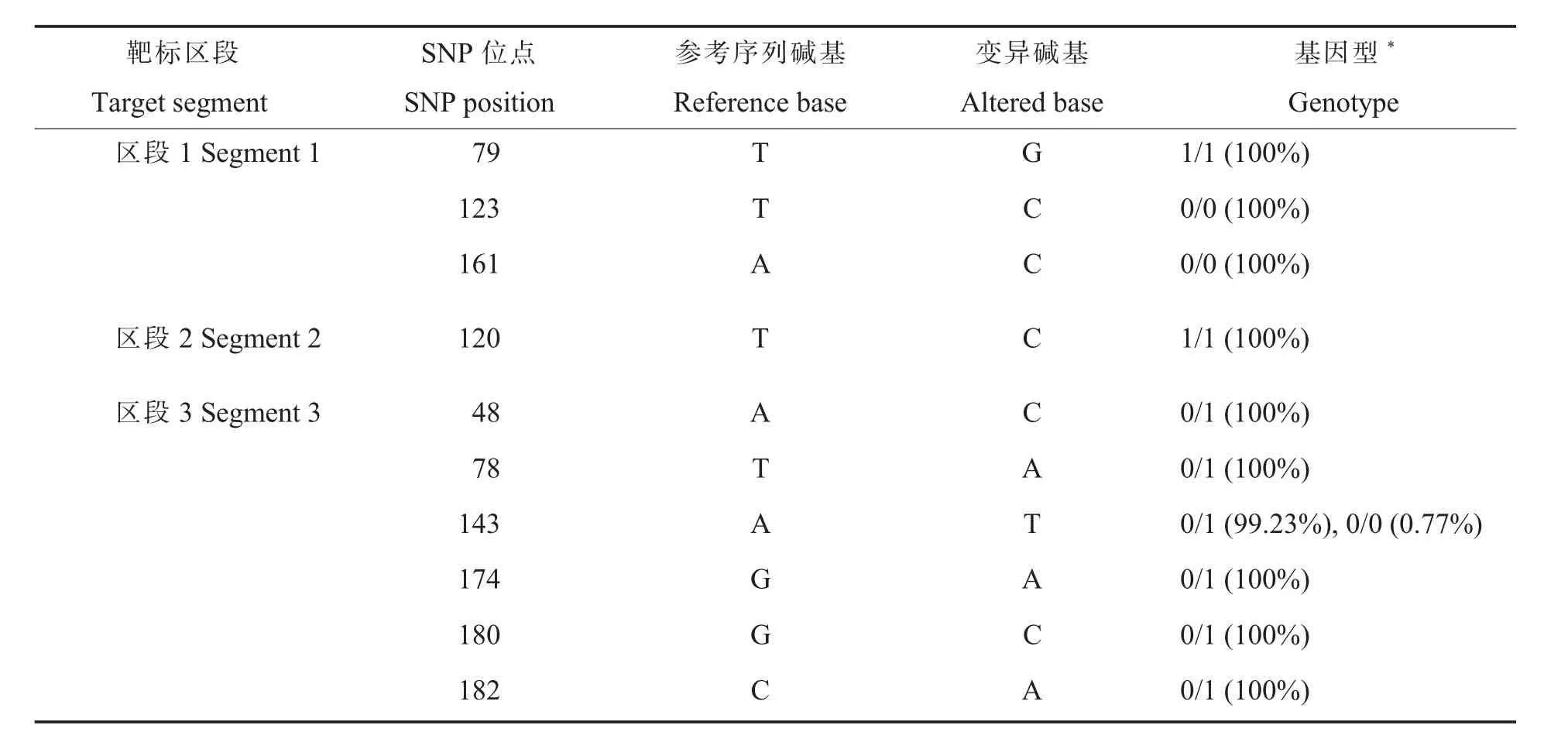
表3 靶标区段作为参考序列的变异检测与分型结果Table 3 Target segment as a reference sequence for variance detection and typing results
2.2 关键参数优化与分析
根据棉花数据库BLAST 结果, 靶标区段3 的同源序列位于D12 染色体47 602 597~47 602 836 bp, 两者相似性达96.28%(图3),BWA 软件比对时无法区分区段3 及其同源序列(同源区段3), 因此考虑将区段3 和同源区段3同时作为参考序列。
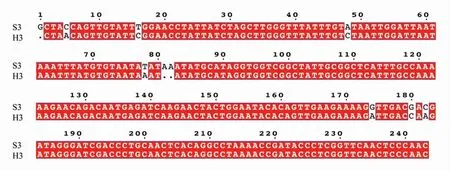
图3 区段3 与同源区段3 的BLAST 比对情况Fig. 3 BLAST comparison of segment 3 and homologous 3
首先, 将同源区段3 单独作为参考序列,其潜在SNP 位点分析结果与区段3 作为参考序列时的分析结果一致。然后,将区段3 与同源区段3同时作为参考序列分析,比对到区段3 与同源区段3 的读长总数为3 462 670,其中约48%比对到区段3,约52%比对到其同源区段。
随机选取1 个样本,从以区段3 作为参考序列分析的SAM 文件中, 随机选取比对到区段3的部分读长,再从以区段3 与其同源区段3 同时作为参考序列分析的SAM 文件中提取相同数量、 相同序列号的读长进行比对和比较可以看到,只有靶标区段作为参考序列时得到4 个杂合位点(图4A);在参考序列中加入同源区段3 后(图4B),不同亚基因组的读长分开,能够直观判断出SNP 和亚基因组内的多态性即HSV。

图4 不同参考序列部分对比结果比较Fig. 4 Comparison of mapping reads with different reference sequences
利用SAMtools 对区段3 和同源区段3 中目标SNP 位点进行变异分析,区段3 的143 位点r值接近1 即基因型为纯合TT, 与GATK 报表中143 位点基因型一致(表4);而其同源序列对应位点141 位点的r值均接近0 即基因型为纯合AA。
比较使用区段3、同源区段3、区段3 加同源区段3 作为参考序列的GATK 结果:在靶标区段3 单独作为参考序列时, 区段3 的143 位点在大部分材料中被鉴定为杂合TA, 在个别材料中被鉴定为AA, 而把靶标区段与其同源区段同时作为参考序列时,143 位点被正确地鉴定为AA 或TT; 区段3 的另外3 个SNP 其实是亚基因组间的HSV;48 位点SNP 和78 位点SNP 是2 个新的SNP(表4)。
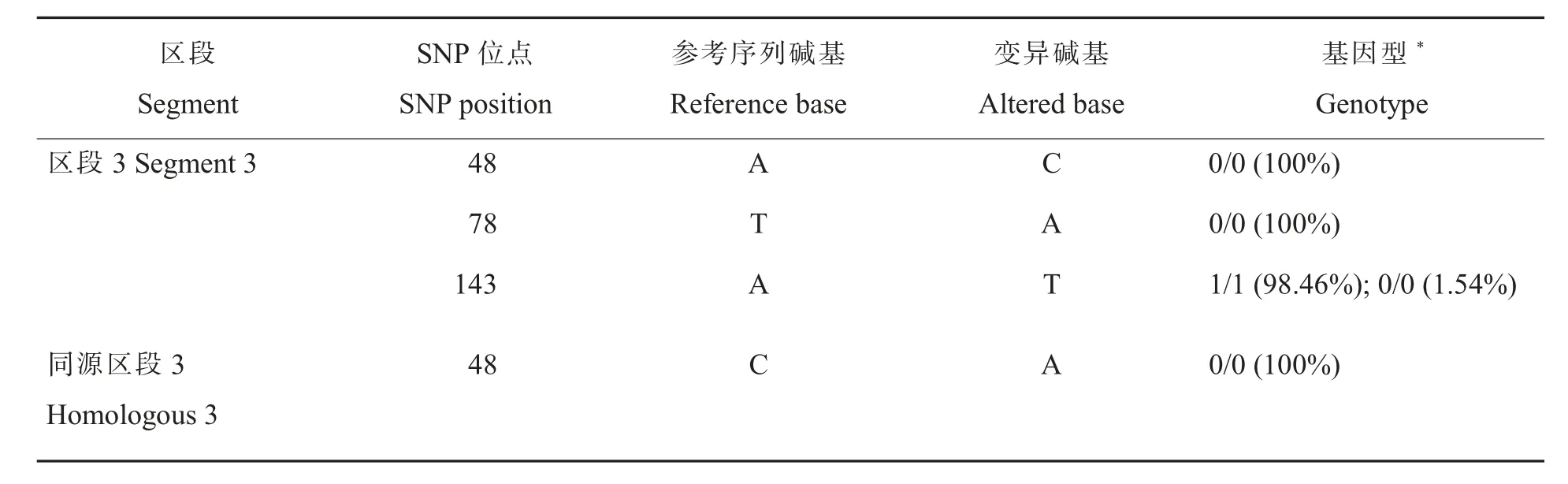
表4 靶标区段和同源区段同时作为参考序列时变异检测与分型结果Table 4 Variation detection and typing results with the target and segment and homologous segment as reference sequences
3 讨论
陆地棉A 亚基因组与D 亚基因组具有极高的同源性[20],存在大量的同源区段干扰SNP 分型。正如Kaur 等[21]所述,在异源多倍体植物中,同源区段间SNP、HSV 和PSV 都有可能存在,比对时由于高度相似的同源位点映射错误,SNP 准确分型较为困难, 而且会增加SNP 的假阳性率,其中关注最多的是区分SNP 与HSV[22]。 PSV(本研究未涉及)大多出现在基因家族成员中,由于横向同源基因的干扰, 使开发SNP 难度大大增加,如陆地棉GhNAC家族[23]。
多倍体植物的分子育种需要精确分型,但常规的技术手段难以区分同源区段[7,24]。基于Sanger测序成本高、通量低,且只能由套峰判断杂合性,而目前最流行的高通量基因分型平台是基于杂交的SNP 阵列和各种使用NGS 的基因分型[25]。特定技术方案有可能解决同源干扰问题,如李杏瑜等[26]利用花生的近缘二倍体和高分辨率溶解曲线(High resolution melting,HRM)方法区分SNP 和HSV。Oliver 等[27]利用生物信息学分析从燕麦(异源六倍体)127 000 个重叠群中筛除HSV,获得了9 448 个候选SNP。 Byers 等[28]尝试在棉花中仅扩增带有SNP 的亚基因组, 虽然成功率低于预期,仍在陆地棉中鉴定出11 834 个SNP。 总之,对不同多倍体的基因分型有不同的最佳策略[29]。 但这些方法或是基于杂交而缺少特异性,或是只适用于没有参考序列的多倍体, 因此需要开发新策略,以高通量方式区分多倍体中部分同源变异与真正等位基因SNP。结合多重PCR 靶向测序[11]与生物信息学方案[14,30],本研究通过对陆地棉样本的常规分析与参考序列调整优化,证明了多倍体中同源区段的存在会影响生物信息学分析与SNP 基因型鉴定,表明多倍体不同亚基因组SNP分型需要个性化的生物信息学方案。
本研究对靶标区段3 潜在SNP 位点常规分析的分型结果为杂合, 但从陆地棉遗传学角度,出现几乎全为杂合子的基因型分布是不合理的;结合多倍体特性,我们猜测并证明了是同源区段干扰导致的这一结果。 由此,本文提出把同源区段与靶标区段同时作为参考序列的方法,区分测序数据中来自不同亚基因组的读长,不仅能得到正确基因型,还能排除HSV 造成的假阳性SNP。区段3 和同源区段3 的分析结果也正好与Clevenger 等[8]提出的“如果亚基因组相差3%左右,则仅允许差异小于3%的reads 比对到正确的亚基因组上”这一结论吻合。
4 结论
通过生物信息学方案优化可有效区分不同亚基因组读长并准确鉴定SNP 的基因型, 提高SNP 分型的准确度, 有助于多倍体品种的检测,同时在多倍体育种方面可以提供DNA 水平上的信息。
致谢:
感谢华中农业大学林忠旭教授提供宝贵的植物材料。
