醋酸乌利司他关键中间体的合成工艺
2021-04-02候美娇董金华
候美娇,董 敏,杨 旭,张 翔*,董金华*
(1.中国医学科学院 北京协和医学院药物研究所 活性物质发现与适药化研究北京市重点实验室,北京 100050;2.沈阳药科大学 基于靶点的药物设计与研究教育部重点实验室,辽宁 沈阳 110016)
醋酸乌利司他(Ulipristal Acetate),化学名为17α-乙酰氧基-11β-[4-(N,N-二甲氨基)苯基]-19-去甲孕甾-4,9-二烯-3,20-二酮,最初是由三角研究所(Triangle)作为一种选择性孕酮受体调节开发。法国HRA制药公司首次推出其作为无保护性交或避孕失败后120 h内的紧急避孕药,并于2009年在美国申请获批上市。2012年,该产品在欧盟获得批准,用于对育龄成年妇女子官肌瘤的中度至重度症状进行术前治疗。2015年,该产品在欧盟获得批准,用于对育龄成年妇女子宫肌瘤的长期管理中的间歇性使用[1]。本品作为一种选择性孕酮受体调节剂(Additional selective progesterone receptor modulators,SPRMs)[2-4],具有直接组织特异性部分孕酮拮抗作用,是目前唯一一个批准用于治疗术前子宫肌瘤的SPRMs药物,同时也是唯一在避孕失败或无防护性生活后120 h内口服以避免怀孕的产品,鉴于其在相关适应症体现出的优势,对其原料药制备工艺进行了深入的研究,具有重要的社会和经济意义。
现有醋酸乌利司他主要的合成路线总结如Scheme 1所示。这些路线存在共性缺欠,有进一步改进提升的空间:一是安全性问题,部分路线使用剧毒试剂(如四氧化锇、氰化钠)(路线1~3);二是经济性问题,现有路线工业化生产成本均较高(路线1[5]反应步骤长,路线2[5]、路线3[6-7]反应条件苛刻,路线4[8]、路线5[9-10]后处理成本高);三是环保性问题,对大气污染比较严重,不符合绿色化学的标准。在综合分析已报道的合成策略后[11-14],以3-缩酮(UPA-O)作为醋酸乌利司他的起始原料,以路线5作为优化起始合成路线(Scheme 2)。
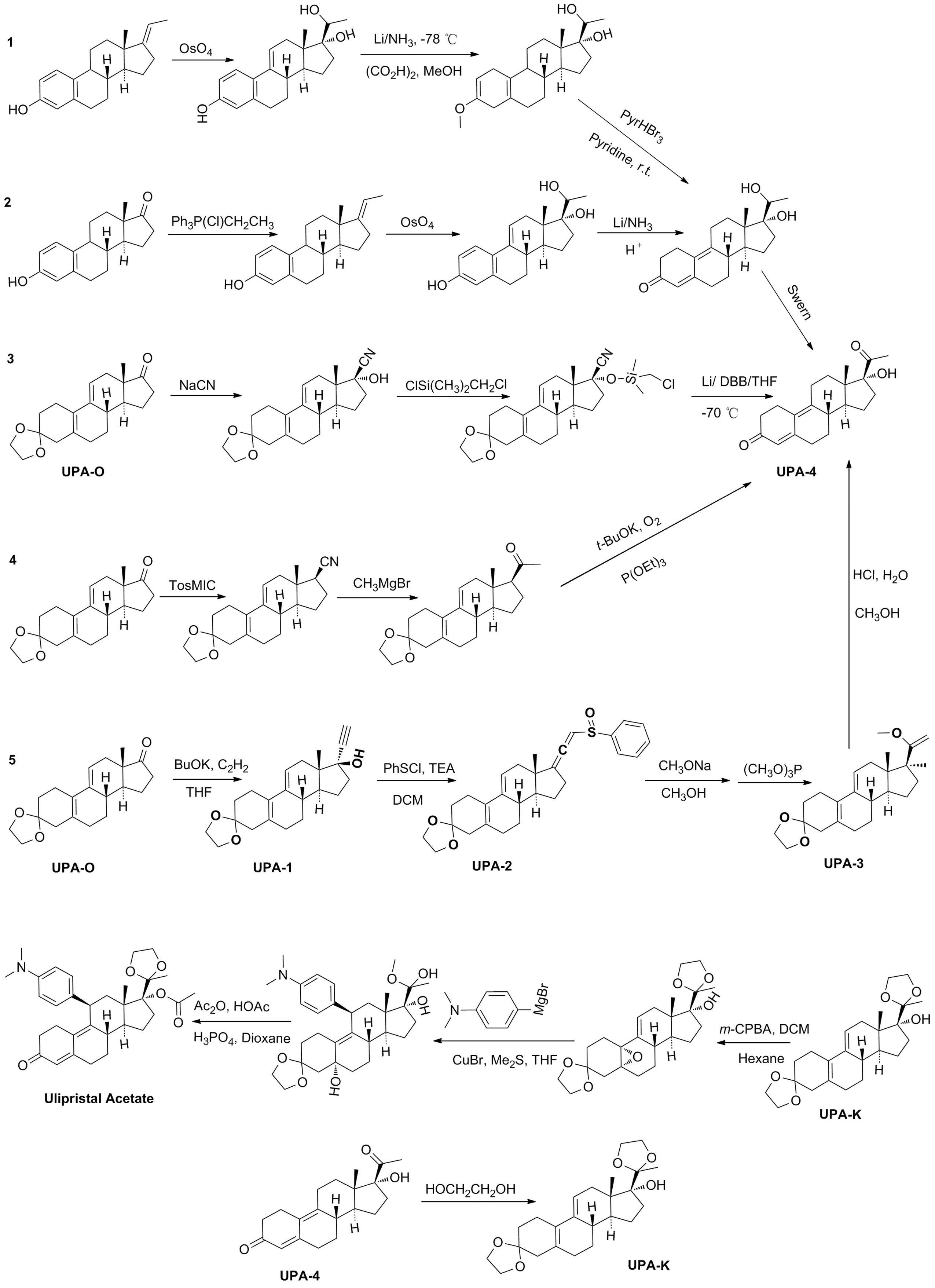
Scheme 1
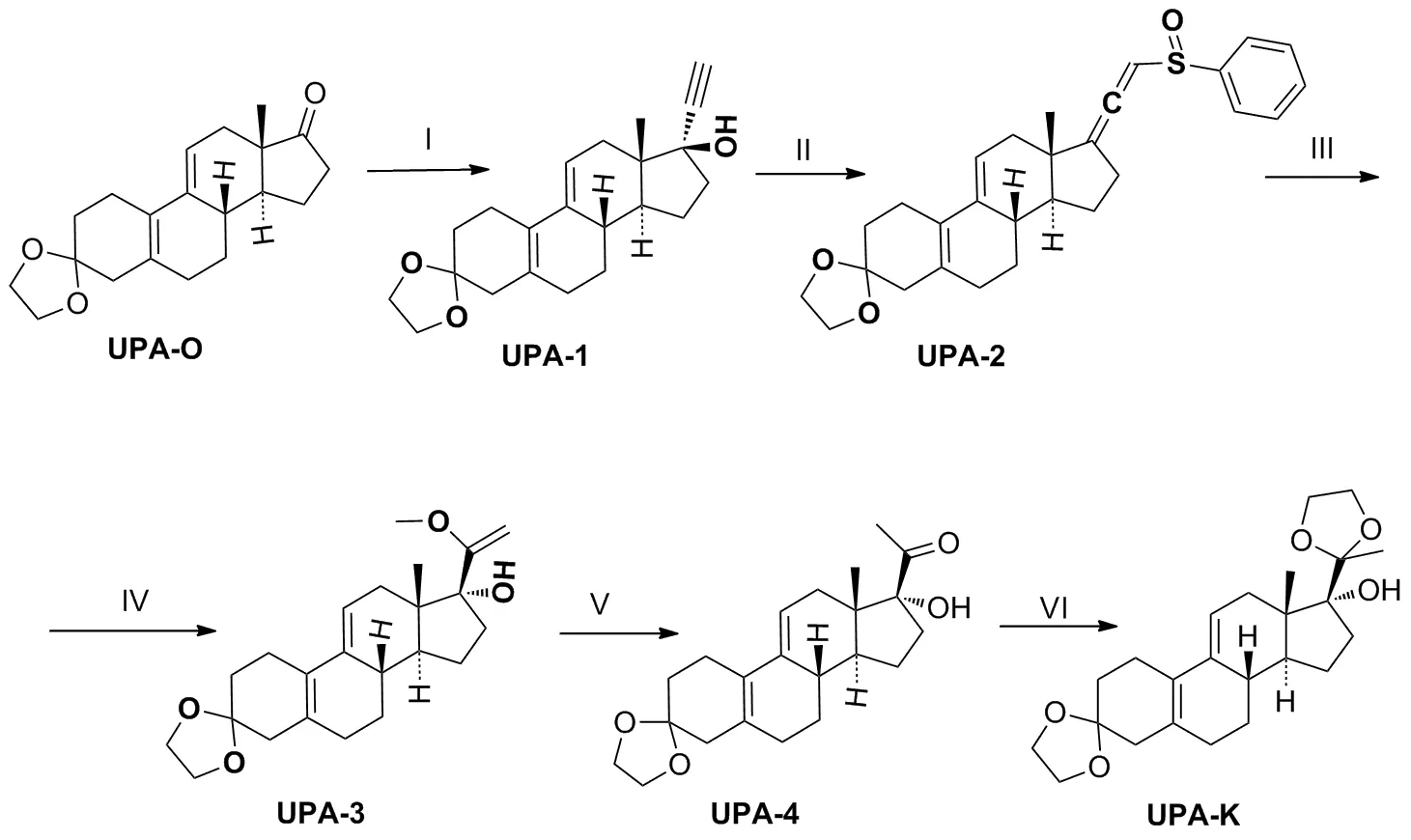
Scheme 2
由于中间体UPA-K对终产物醋酸乌利司他的纯度及收率有着至关重要的影响,因此对此关键中间体制备的每步条件均进行考察,着重于研究可替换汞盐催化炔基水化的Mislow-Evans重排反应条件筛选,实现炔基向乙酰基的顺利转化,以预期得到一条收率较高、符合绿色化学要求,可工业化生产以及相对经济合理的合成路线。
1 实验部分
1.1 仪器与试剂
Yanaco MP-J3型毛细管熔点仪;Mercury-400型核磁共振仪(DMSO-d6或CDCl3为溶剂,TMS为内标),Biotage Isolera one型快速制备色谱;Thermo Exactive Orbitrap型质谱仪。
所用试剂均为分析纯或化学纯。
1.2 合成
(1) 3-亚乙二氧基-17α-乙炔基-17β-羟基-19-去甲孕甾-5(10),9(11)-二烯(UPA-1)的合成
称取叔丁醇钾3.5 g(31.2 mmol)溶解于60 mL 干燥THF中,室温下通入干燥乙炔30 min(浓硫酸洗气),降温至-5 ℃后继续通乙炔1 h,加入化合物UPA-O6.5 g(20.6 mmol),并继续通乙炔2 h(TLC监测)。向反应液中加入饱和氯化铵溶液60 mL,搅拌1 min,减压除去THF至溶液呈粘稠状,搅拌下加入冰水50 mL,并在冰浴中搅拌2 h,有淡黄色固体析出,过滤,滤饼用水(3×5 mL)洗涤,于30 ℃真空干燥得类白色固体UPA-16.57 g(19.31 mmol),收率96.6%,m.p.155~157 ℃(145~148 ℃[15]);1H NMR(DMSO-d6,400 MHz)δ:0.71(s,3H),1.02~2.57(m,18H),3.33(s,1H),3.86(s,4H),5.53(t,1H),5.39(s,1H);MS(ESI)m/z:Calcd for C22H28O3{[M+H]+}340.2038,found 340.2105。
(2) 3-亚乙二氧基-21-苯基亚磺酰基-19-去甲孕甾-5(10),9(11),17(20),20-四烯(UPA-2)的合成
依次称取1.83 mL(13.22 mmol)三乙胺和0.84 mL冰醋酸(13.35 mmol)室温下搅拌1 h,然后加入UPA-11.5 g(4.41 mmol)溶解于10 mL干燥DCM中,氩气保护下,于-5 ℃缓慢滴加含苯次磺酰氯0.536 mL(4.62 mmol)的5 mL干燥DCM中,搅拌30 min(TLC监测)。加入水15 mL和甲醇5 mL搅拌30 min,分液,用DCM(3×10 mL)萃取水相,合并有机相,依次用水(3×30 mL)、饱和食盐水(30 mL)洗,无水硫酸钠干燥、过滤,减压除去DCM得黄色胶状固体UPA-22.31 g,不经纯化直接用于下步反应,m.p.185~187 ℃(176~180 ℃[9-10]);1H NMR(DMSO-d6,400 MHz)δ:0.78~0.80(d,3H),1.12~2.61(m,18H),3.82(s,4H),5.46(s,1H),6.44(s,1H),7.51~7.58(m,5H);MS(ESI)m/z:Calcd for C28H32O3{[M+H]+}448.2072,found 448.2137。
(3) 3-亚乙二氧基-17α-羟基-20-甲氧基-19-去甲孕甾-5(10),9(11),20-三烯(UPA-3)的合成
依次称取UPA-22.31 g(4.41 mmol)和甲醇钠0.716 g(13.25 mmol)溶解于15 mL无水甲醇中,氩气保护下,65 ℃加热回流2.5 h(TLC监测)。加入二乙胺4.56 mL(44.13 mmol),继续反应3.5 h后加入1 mol/L柠檬酸水溶液50 mL和DCM 30 mL,搅拌30 min,分液,有机相依次用1 mol/L柠檬酸水溶液(50 mL)、水(30 mL)、饱和食盐水(30 mL)洗涤,无水硫酸钠干燥、过滤,30 ℃减压除去DCM得黄色胶状固体UPA-32.43 g,m.p.128~132 ℃(128~132 ℃[9-10]);MS(ESI)m/z:Calcd for C23H32O4{[M+H]+}372.2301,found 372.2367。
(4) 3-亚乙二氧基-17α-羟基-19-去甲孕甾-5(10),9(11) -二烯-20-酮(UPA-4)的合成
将上步所得UPA-32.43 g(4.41 mmol),加入15 mL甲醇溶解,降温至0 ℃,搅拌下加入1 mol/L盐酸(10 mL),有大量白色固体析出(TLC监测)。过滤,滤饼用甲醇(3×3 mL)洗涤得白色固体UPA-41.404 g,收率为88.9%,m.p.197~202 ℃(140 ℃[9-10]);1H NMR(CDCl3,400 MHz)δ:0.68(s,3H),0.81~2.86(m,18H),2.22(s,3H),3.96(s,4H),5.55(s,1H);MS(ESI)m/z:Calcd for C22H30O4{[M+H]+}358.2144,found 358.2208。
(5) 3,20-二(亚乙二氧基)-17α-羟基-19-去甲孕甾-5(10),9(11)-二烯(UPA-K)的合成
称取UPA-41.0 g(2.79 mmol),溶解于5 mL DCM中,然后向其中加入乙二醇5 mL(89.86 mmol),原甲酸三甲酯1.22 mL(11.16 mmol)及对甲苯磺酸0.048 g(0.27 mmol),60 ℃下加热搅拌30 min(TLC监测)。依次加入饱和碳酸氢钠溶液30 mL,DCM 20 mL,搅拌15 min,分液,有机相用饱和NaCl溶液(3×15 mL)洗涤,无水硫酸钠干燥、过滤,30 ℃减压除去DCM得透明胶状固体,用5 mL冰甲醇打浆,有白色固体析出,过滤,滤饼用冰甲醇(3×3 mL)淋洗,30 ℃真空干燥6 h得白色固体UPA-K0.98 g,收率87.3%,m.p.175~177 ℃(170~172 ℃[11]);1H NMR(CDCl3,400 MHz)δ:0.79(s,3H),1.26~2.74(m,18H),1.40(s,3H),3.84~4.06(m,8H),5.56(s,1H);MS(ESI)m/z:Calcd for C25H38O5{[M+H]+}402.2719,found 402.2463。
2 结果与讨论
2.1 合成工艺考察
(1) 3-亚乙二氧基-17α-乙炔基-17β-羟基-19-去甲孕甾-5(10),9(11)-二烯(UPA-1)的合成工艺
主要考察了碱的用量对反应收率及时长的影响,结果如表1所示。由表1可知,适当增加BuOK用量可以缩短反应时长,收率提高,但增加至2.0 eq.时收率并没有进一步提高,反应时长也没有相应缩短,所以该步反应条件为:溶剂为THF,nUPA-O/nBuOK=1/1.5。
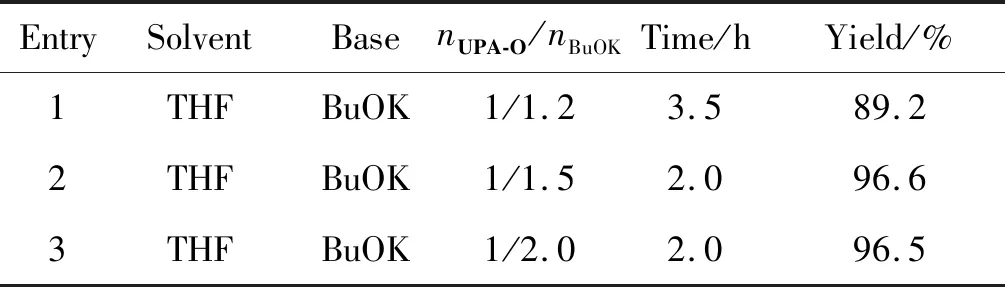
表1 UPA-1合成优化条件的考察Table 1 Optimal conditions for synthesis of UPA-1
(2) 3-亚乙二氧基-21-苯基亚磺酰基-19-去甲孕甾-5(10),9(11),17(20),20-四烯(UPA-2)的合成工艺
该步反应起始研究收率为67.2%(Entry 1),TLC监测原点附近有一个明显的杂质点,推测可能是由于过量的苯次磺酰氯可继续与联烯亚砜化合物UPA-2反应产生杂质,而体系中游离的三乙 胺可以促进该副反应的进行,导致该步收率降低和后处理困难。将三乙胺制成三乙胺弱酸盐参与反应,保证体系中没有游离的三乙胺;降低反应温度减少副反应的发生;改变反应溶剂;减少苯次磺酰氯的用量。具体优化条件如表2所示。

表2 UPA-2合成优化条件的考察Table 2 Optimal conditions for synthesis of UPA-2
实验结果显示:三乙胺醋酸盐的效果要优于三乙胺;低温可以降低副反应的发生,但也延长了反应时间,增加了工业成本;相同条件下DCM的反应效果要优于THF。综合考虑最后决定该步的反应条件为以三乙胺醋酸盐参与反应,温度为-5 ℃时,nUPA-1/nTEA/nPhSCl=1/3.0/1.05。
(3) 3-亚乙二氧基-17α-羟基-20-甲氧基-19-去甲孕甾-5(10),9(11),20-三烯(UPA-3)的合成工艺该步反应主要考察溶剂和Thiophile试剂种类对反应收率的影响。优化条件如表 3所示。

表3 UPA-3合成优化条件的考察Table 3 Optimal conditions for synthesis of UPA-3
实验结果显示:甲醇为溶剂时反应效果最好;Thiophile试剂中三苯基膦(PPh3)效果最好,哌啶、二乙胺收率略低于三苯基膦;但三苯基膦工业上后处理困难,哌啶、二乙胺可以通过调节反应液PH去除,且二乙胺成本低于哌啶,所以最终选定反应条件是以甲醇为溶剂,温度为65 ℃时,nUPA-2/nCH3ONa/n二乙胺= 1/3.0/10.0。
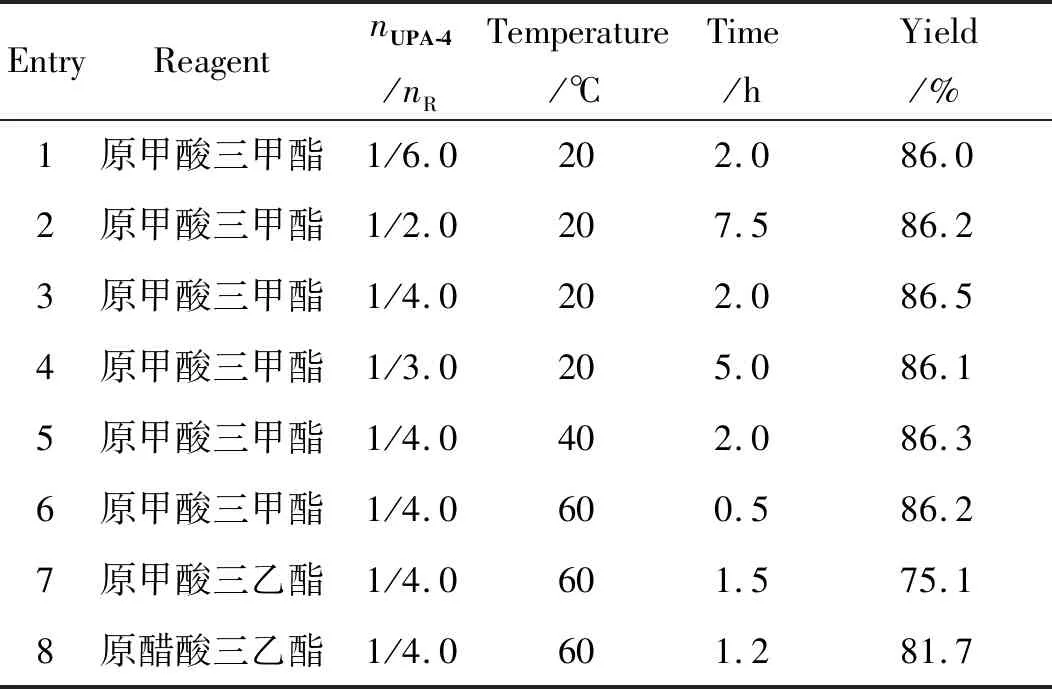
表4 UPA-K合成优化条件的考察Table 4 Optimal conditions for synthesis of UPA-K
(4) 3-亚乙二氧基-17α-羟基-19-去甲孕甾-5(10),9(11) -二烯-20-酮(UPA-4)的合成工艺
该步反应收率波动范围较大,产生极性较大的杂质,是由于反应时间过长,部分产物在盐酸作用下脱掉缩酮保护基使收率不稳定。实验证实,将上一步未经纯化的产物减压浓缩后,0 ℃下搅拌加入预冷的甲醇与1 mol/L盐酸,立刻有白色固体析出,确证为UPA-4。
(5) 3,20-二(乙二氧基)-17α-羟基-19-去甲孕甾-5(10),9(11)-二烯(UPA-K)的合成工艺
由表中数据可知,使用原甲酸三甲酯时,收率最高;升高温度可以缩短反应时间,原甲酸三甲酯用量降低到4.0 eq.时不会延长反应时间,确定最终条件为,温度为60 ℃,以原甲酸三甲酯为反应试剂,nUPA-4/nR=1/4.0。
2.2 UPA-K绝对构型
通过单晶衍射确定了UPA-K的绝对构型,UPA-K为斜方晶系,属于P21212空间群,晶胞参数为a=19.5823(5) Å,b=14.4786(3) Å,c=7.20930(17) Å ;α=90.00°,β=90.00°,γ=90.00°,U=2044.00(8) Å3,T=109.65(10),Z=4,μ(Cu Kα)=0.724,wR(F2)=0.0971。
主要对醋酸乌利司他关键中间体(UPA-K)的合成路线进行了优化研究。通过对已有合成策略综合分析、选择、优化,最终确定以3-缩酮(UPA-O)为原料,经亲核取代、亲核加成、Mislow-Evans重排,醚键断裂、烯醇式互变、缩酮保护等5步反应,制备目标产物UPA-K的工艺路线。优化后的路线成本低、收率较高且稳定、操作简单、符合绿色化学标准,适合工业放大化生产。整条路线总收率74.8%,较已有路线报道收率显著提高,为醋酸乌利司他的工业放大化生产提供新的选择。
