前列腺间质状态在良性前列腺增生的发生发展中的转录组学分析
2021-03-03徐向来杨正青王佳骏朱延军
徐向来, 杨正青, 王佳骏, 朱延军*
1. 复旦大学附属中山医院泌尿外科,上海 200032 2. 上海杨思医院泌尿外科,上海 200126
良性前列腺增生症(BPH)是造成老年男性膀胱出口梗阻的常见原因,其引起的下尿路症状(LUTS)大大降低了患者生活质量[1]。BPH的患病率随着年龄增长逐渐增加,50%的50岁以上男性存在组织学上的前列腺良性增生[2]。目前,BPH的治疗药物主要包括5α还原酶抑制剂、α受体阻滞剂和5型磷酸二酯酶抑制剂。然而,仍有大量患者患有难以通过药物改善的LUTS症状及并发症,出现反复的尿潴留[3-4]。这些患者需手术干预,例如经尿道前列腺切除术(TURP)和激光剜除前列腺(HoLEP)[5]。
BPH的组织学改变是基质细胞和上皮细胞的过度生长,主要发生于前列腺的移行带。BPH的发病与雄激素信号通路的改变、间质活化和炎症免疫反应[1-2]相关。雄激素信号通路激活前列腺基质相关的信号通路,造成增生相关生长因子的表达,进一步造成间质细胞的增殖和分化[6-7]。前列腺间质改变在BPH发生发展中起重要作用,而局部缺血缺氧可能是这种间质病变的诱因。BPH相关的前列腺炎症也被认为是BPH的重要因素[8-9]。尽管已有研究团队对前列腺间质增生、变化在BPH中的机制进行过探索,但是尚无转录组水平的深入研究。
转录组学技术已广泛用于人类疾病的研究。几十年前,就有团队使用芯片进行了BPH表达谱的初探[10-11]。近年来,基因组学、转录助学和表观基因组学分析等,已被用于BPH研究[12-14]。这些研究在一定程度上揭示了BPH发生发展的生物学过程。研究人员发现了2种分子分型的BPH亚组,代表不同的生物学行为[12]。另外,Middleton等[13]发现骨形态发生蛋白5(BMP5)等基因的表达也与BPH疾病进展有关。本研究拟从转录组学入手,观察BPH患者前列腺间质组织成分变化,并分析正常前列腺和BPH患者前列腺标本的间质相关基因差异及特征。
1 材料与方法
1.1 研究人群和数据采集 研究工作流程如图1。本研究搜索了Gene Expression Omnibus(GEO)以获取BPH的转录组数据,共收集了34个来自GEO数据集(GSE119195、GSE7307、GSE101486)的BPH样品和13个正常前列腺样品的转录组测序数据(表 1)。其中,GSE119195的BPH标本来自全膀胱切除标本中的前列腺组织,正常前列腺组织来自脑死亡患者捐赠的标本。GSE7307的BPH和正常前列腺组织,均来自脑死亡患者捐赠的标本。GSE101486的BPH标本来自TURP标本,与前列腺剜除标本。所有BPH标本均为前列腺移行带标本,均经过组化病理分析排除前列腺恶性肿瘤;所有正常对照标本,均经过病理分析排除BPH及前列腺恶性肿瘤。另一个数据集(GSE132714)含18个BPH样本(来自TURP标本)和4个正常前列腺样本(来自T2a前列腺肿瘤标本的对侧正常移行带)用作验证数据集。R软件(版本4.0.2;https://www.r-project.org/)用于数据分析。sva 程序包用于去除批次效应[15]。

表1 GEO数据库中调取的BPH及正常组织的个数

图1 分析流程图
1.2 样本微环境分析 定量基质评分采用ESTIMATE算法[16],每个样本的微环境细胞组成计算采用加州大学旧金山分校开发的xCell算法[17]。
1.3 加权基因共表达网络分析(WGCNA) WGCNA是利用基因表达数据构建无标度网络的系统生物学方法[18]。本研究通过WGCNA分析基因组和表型之间的关联,发现可能与BPH发育有关的潜在生物标志物基因。将基因表达矩阵和表型数据构建为输入数据。使用R软件中的“WGCNA”软件包执行分析。
1.4 功能富集分析 通过“Limma”包计算BPH组织和正常前列腺组织的差异表达基因(differentially expressed gene, DEG),采用t检验进行统计分析,将P<0.001和表达倍数绝对值>2的基因视为差异表达基因[19]。使用Metascape进行基因本体论(gene onotology, GO)分析和Kyoto encyclopedia of genes and genomes(KEGG)富集分析。
1.5 蛋白质相互作用网络分析(PPI) 使用search tool for the retrieval of interacting genes/proteins (STRING)工具进行蛋白互作网络分析。此外,对PPI数据,进一步使用Cytoscape 3.7.2版中的cytoHubba插件进行关键基因(hub gene)的筛选。使用以下4种算法计算hub基因:maximum neighborhood component (MNC)、maximal clique centrality (MCC)、maximum neighborhood component (DMNC)、degree。根据分值的高低筛选出排名前15的关键基因。
1.6 主成分分析(PCA) PCA是利用降维的方法,将多个变量转化降为成为几个综合的变量(主成分),每个成分之间互不相关。这些主成分能在信息不重叠的情况下,较好地反映原始变量的绝大部分信息。因此,PCA是一种能够在最大化降低保持数据信息损失的原则下,对高维变量空间进行降维处理的线性映射方法。本研究使用PCA评估筛选的PCA基因是否能够良好地区分BPH标本及正常前列腺标本。
1.7 LASSO回归方法 LASSO回归的原理是在最小二乘的基础上,增加一个惩罚项来对估计系数进行压缩。当系数在惩罚项的作用下无限趋于0时,就可选择出对因变量影响较大的自变量和对应的系数估计值。LASSO回归常用于处理存在多重共线性的样本数据。参数λ决定了LASSO回归惩罚项的压缩程度,λ越大则惩罚项的影响力越大。通过LASSO回归,可使最终纳入模型的变量均为与因变量显著相关(P<0.05)且考虑变量共线性问题的自变量集合。LASSO回归可避免参数过度拟合,适合可给出一个具备优良性能但是自变量个数相对较少的诊断模型。
2 结 果
2.1 BPH组织和正常前列腺组织中间质细胞占比不同 本研究从GEO数据库下载了3个数据集,去除批次效应,合并成为包含了34个BPH和13个正常前列腺样品的转录组数据和表型数据的探索数据集(图1)。通过ESTIMATE程序包计算了在BPH和正常前列腺组织中的基质评分发现,BPH组织中间质评分显著高于正常前列腺组织(P=0.025,图2)。结果表明,BPH组织中的间质成分占比显著多于正常前列腺组织。为了进一步研究BPH发生发展过程中的间质变化,本研究使用xCell算法对BPH和正常前列腺组织样本中的间质细胞进行详细计算(图3A、图3B)。目前,尚无研究团队对BPH组织的间质组成进行分型分析,特别是不同类型或表型的间质细胞。通过xCell算法发现,BPH样品中的成纤维细胞、周细胞显著增加(P均<0.05)。相反,内皮细胞、肌细胞、成骨细胞和平滑肌细胞组织减少(P均<0.05,图3A)。本研究还发现在BPH组织中成纤维细胞/平滑肌细胞的比例显著升高(P=0.001 8,图3C)。从转录组数据可推断,成纤维细胞或平滑肌细胞的升高,也是BPH组织的特征之一。

图2 BPH组织中间质评分显著高于正常前列腺组织

图3 xCell分析BPH和正常前列腺组织样本中的间质细胞A-B:BPH样品中的成纤维细胞、周细胞显著增加。相反,内皮细胞、肌细胞、成骨细胞和平滑肌细胞组织减少;C: BPH组织中成纤维细胞/平滑肌细胞的比例显著升高(P=0.001 8)。
2.2 WGCNA模块分析与BPH及间质相关的关键模块 WGCNA构建的输入表达谱矩阵和表型矩阵包含常见的14 24个基因和34个样品的表型数据。输入的表型数据包括间质评分、成纤维细胞、平滑肌细胞、内皮细胞。根据表达谱数据,计算每个样品中每个基因的方差。选择标准偏差大于1.2的基因并进一步聚类所有样品,并标注样本特征(图4)。所有样品均符合WGCNA的筛选标准,未排除样品。使用GCNA包进行构建权重共表达网络,使用分析包自动选择的软阈值计算得到软阈值β=18作为软阈值进行后续分析。确定软阈值后,通过动态剪切树法进行模块初步识别并合并相似模块,设置每个基因网络模块最少的基因数目为30。总共获得18个模块,其中灰色模块是不能聚合到其他模块中的基因的集合(图5A)。

图4 WGCNA聚类所有样品,标注样本特征,切割合并模块A:WGCNA算法对样品进行聚类,未发现需要剔除的样本;B:以0.2相异性系数为阈值合并相关的基因模块。
根据每个模块的特征向量,计算这些模块和每个输入表型(免疫评分、成纤维细胞、平滑肌细胞、内皮细胞)之间的相关性。对富集到的模块进行GS分布预测。每个模块中每种表型的GS分布可显示表型和基因之间的相关性(图5B、图6)。结果提示,volet、darkgrey、lightyellow、darkorange、purple、darkolivegreen、pink、greenyellow、lightcyan1模块和间质评分、成纤维细胞、平滑肌细胞相关。因此,提取这9模块所包含的基因作为进一步分析。
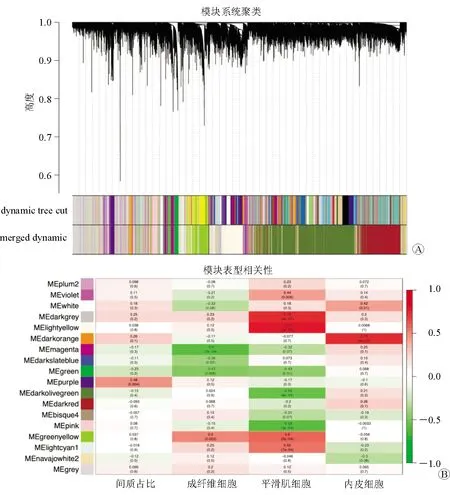
图5 WGCNA合并模块,对表型信息进行相关性分析A-B:通过动态剪切方法确定模块后,依次计算每个模块的特征向量,然后聚类模块并将更近的模块合并到新模块中,共获得18个模块,其中灰色模块是不能聚合到其他模块中的基因的集合。
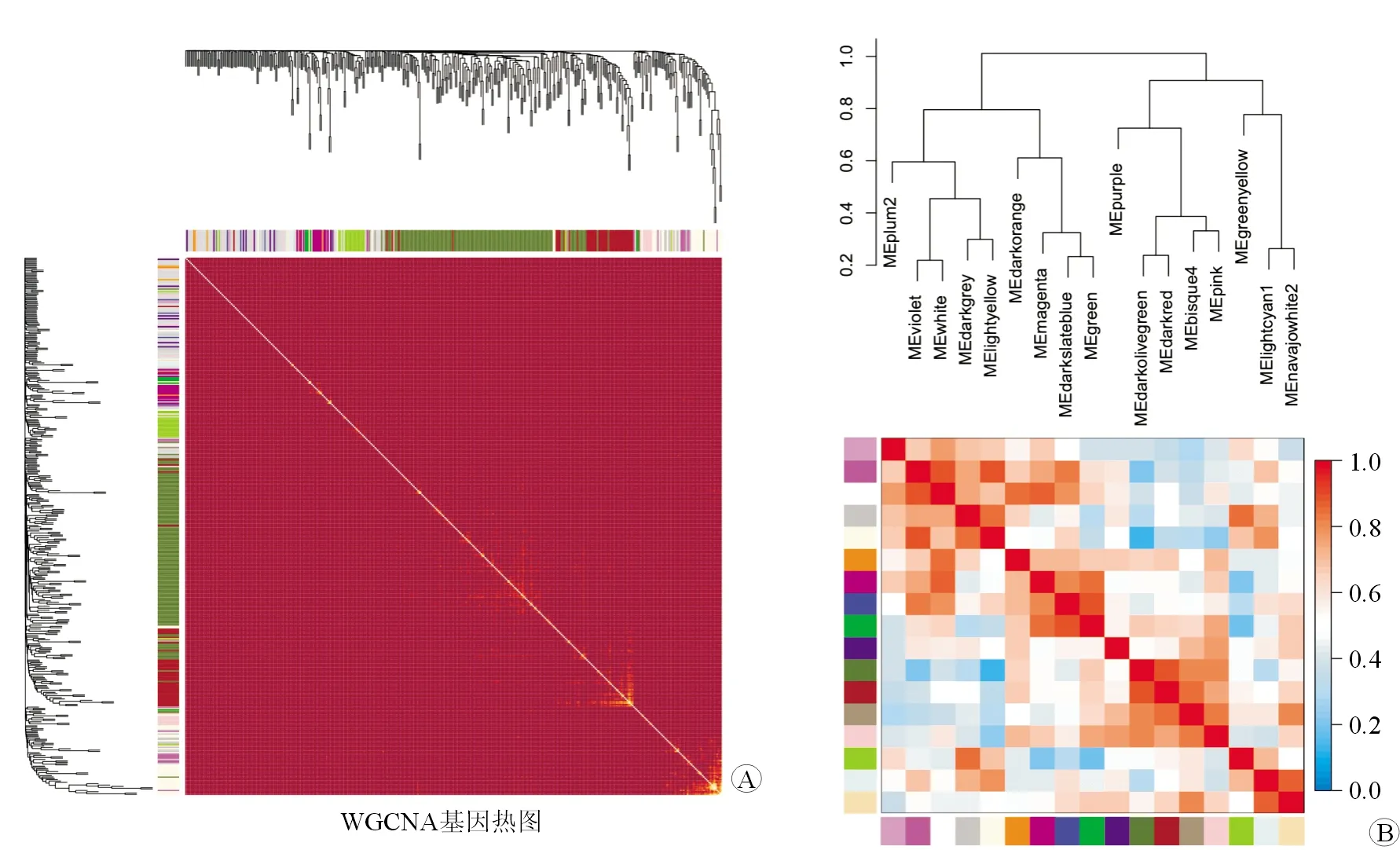
图6 WGCNA基因画拓扑重叠热图和特征向量基因A:随机选择400个基因画拓扑重叠热图,行和列都表示单个基因,深黄色表示高度的拓扑重叠;B:计算每个模块的特征向量基因,绘制特征向量基因临近热图,代表该模块内基因表达的整体水平。
2.3 前列腺间质增生相关的关键基因 使用“limma”程序包,对BPH和正常前列腺样本的表达谱数据进行了差异表达分析。|logFC|≥1且P<0.05为阈值,共筛选出98个上调基因和204个下调基因,DEGs见火山图(图7A)。将DEGs与上一小节中的9模块的基因取交集。结果发现violet、darkgrey、lightyellow、darkorange、lightcyan1模块与差异基因并无交集,因此剔除了这5个模块。从其余4模块purple、greenyellow、pink、darkolivegreen中,共提取到122个基因(图7B)。进一步的GO富集分析发现多个纤维增生,肌细胞分化、增殖,细胞外基质等功能模块富集(图8A)。KEGG富集分析发现,筛选的基因集在Hippo、丝裂原活化蛋白激酶(MAPK)、哺乳动物雷帕霉素靶点(mTOR)等通路富集(图8B)。
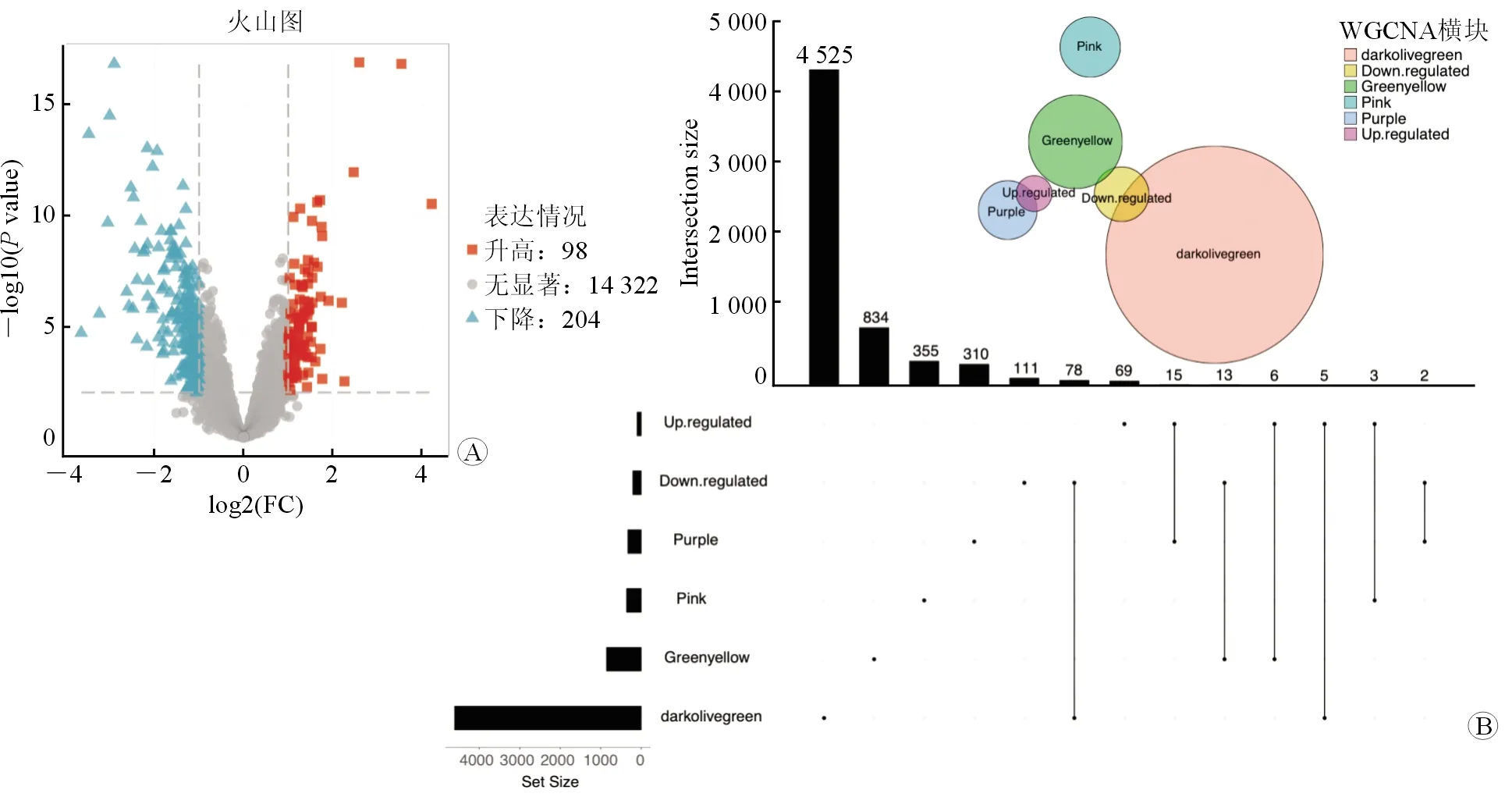
图7 联合差异基因进一步筛选WGCNA前列腺间质增生相关基因A:BPH组织和正常前列腺组织的差异表达基因;B,差异表达基因及WGCNA的purple、greenyellow、pink、darkolivegreen模块联合分析。

图8 WGCNA前列腺间质增生相关差异基因的富集分析A: GO富集分析发现,多个纤维增生,肌细胞分化、增殖,细胞外基质等功能模块富集;B:KEGG富集分析发现,筛选的基因集在Hippo、MAPK、mTOR、TGF-β等通路富集。
GO和KEGG富集分析的结果进一步证明,BPH组织中的成纤维细胞相关通路激活,诱导成纤维细胞增生、分化,这一过程可能参与了BPH的发生和发展。
本研究将筛选所得的122个基因输入STRING数据库,构建PPI网络(图9A)。并使用Cytoscape的cytoHubba插件对hub基因进行筛选。使用了MCC、DMNC、MNC、Degree4算法计算top15的hub基因,取交集获得与前列腺间质增生的相关hub基因(图9B)。通过PCA发现,12个hub基因能够很好的区分前列腺组织和BPH组织,验证了这12个基因在前列腺增生发生中可能发挥了重要作用(图10A~10C)。

图9 蛋白互作网络分析及关键基因筛选A:本研究筛选所得的122个基因构建PPI网络;B:使用Cytoscape的cytoHubba插件对hub基因进行筛选。MCC、DMNC、MNC、Degree 4种算法计算top15的关键基因,取交集获得与前列腺间质增生的相关关键基因。

图10 hub基因的表达和主成分分析A:12个hub基因在前列腺组织和BPH组织的表达热图;B:12个hub基因的蛋白互作网络关系;C:12个hub基因的主成分分析能够很好的区分前列腺组织和BPH组织。
2.4 hub基因与前列腺的间质状态相关 获得了hub基因后,本研究进一步分析了这12个差异基因与增生前列腺间质状态的关系(图11)。肌球蛋白轻链1(MYL1)、肌球蛋白轻链2(MYL2)、伴肌动蛋白(NEB)、Kirsten大鼠肉瘤病毒癌基因同源物(KRAS)、Snail家族转录抑制子2(SNAI2)等与间质状态正相关。其余基因未有统计学意义可能与较少的样本量有关。相反,人肌球蛋白重链3(MYH3)、人肌球蛋白重链2(MYH2)等与间质状态负相关。
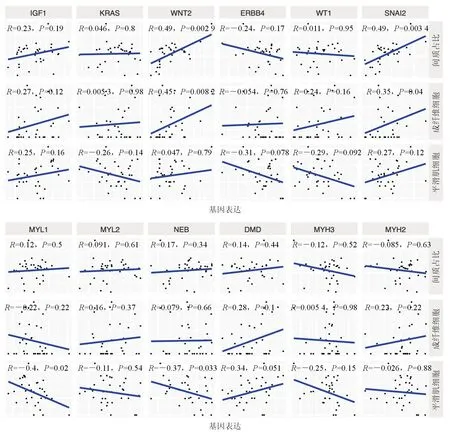
图11 关键基因与增生前列腺间质状态的相关性分析
2.5 BPH间质相关标记基因和模型的建立和验证 本研究分析了12个hub基因在BPH组织和正常前列腺组织中的表达量(图10A)发现,WNT2、胰岛素样生长因子1(IGF1)、SNAI2在BPH组织中表达较高。目前,在BPH研究领域,如何鉴定BPH标本或正常前列腺标本是一个突出的难题。前列腺体积较大,肉眼观察难以直接分辨病变组织与正常组织,在收集患者的手术标本以后,研究者往往难以明确标本的具体病例性质。因此,本研究尝试建立BPH的基因诊断模型,利用LASSO回归从12个hub基因中进行筛选,最终筛选出4个变量(图12A、12B)。分别为NEB、DMD、IGF1、KRAS回归系数分别为-0.980 750、-4.091 432、4.339 909、-2.959 698。利用建立的模型在验证集中进行验证(图12C)显示,该模型能够很好地对BPH组织进行基因诊断分类(AUC=0.889)。

图12 BPH基因模型的建立和验证A:LASSO回归从12个hub基因中进行筛选,最终筛选出NEB、DMD、IGF1、KRAS建立模型,回归系数分别为-0.980 750、-4.09 143 2、4.339 909、-2.959 698;B:利用建立的模型在验证集中进行验证。
3 讨 论
BPH是老年人膀胱出口梗阻的主要原因,能够引起LUTS症状,影响生活质量,但BPH发生的分子生物学机制尚不明确。本研究应用了生物信息学技术,通过分析BPH组织和正常前列腺组织的转录组数据,确认了BPH组织的间质状态,得到了一个由4个DEG组成的标记基因,同时在验证数据集中得到了验证(AUC=0.889)。这可能为BPH的分子学诊断提供了新的思路,也为进一步研究BPH的发生发展的分子生物学机制提供了新的方向。
研究[20]表明,BPH患者存在不同程度的间质成分改变,并和LUTS症状的相关。但尚无研究团队对BPH间质的细胞组成进行精准分析,并对其进行基因分型。BPH患者前列腺间质组织中胶原成分与肌纤维比例低于正常前列腺组织,该比例与LUTS症状正相关。而胶原蛋白正是由成纤维细胞分泌,并受TGF-β等通路调节。这从侧面验证了本研究的结论。同时,慢性炎症也是BPH的重要驱动因素[21]。有研究[22]分离出BPH患者前列腺的原代间质细胞与永生化的前列腺上皮细胞株BPH-1进行体外共培养,能够加快BPH-1的增殖,而从正常前列腺分离出的间质细胞与BPH-1细胞系体外共培养则不能刺激BPH-1的增殖,这说明BPH的间质细胞能够外分泌特定的生长因子刺激前列腺上皮增生。另一项研究[23]显示,前列腺间质细胞能够分泌多种生长类细胞因子如VEGF、成纤维细胞生长因子(FGF)、TGF-β等细胞因子,刺激前列腺间质细胞增生,特别是成纤维细胞,诱导成纤维细胞分泌胶原蛋白。而与之相反,前列腺上皮细胞不能分泌这类细胞因子。因此,前列腺间质状态改变在BPH发生发展中起重要作用。
WGCNA是利用基因表达数据和表型数据构建无标度网络。它能寻找协同作用的基因模块,探索基因网络和感兴趣的疾病表型之间的关系,及网络中的中枢基因。WGCNA分析方法已成为疾病研究领域的一种重要生物信息学分析手段,它侧重于基因共表达模块和表型特征之间的关联,因而具有相对较高的可靠性和生物学意义。本研究通过WGGCNA方法构建共表达模块,分析了与BPH组织中间质评分、成纤维细胞、平滑肌细胞相关的表达模块,确定了4个与BPH间质评分、成纤维细胞、平滑肌细胞显著相关的共表达模块,用于检测BPH转录组与BPH组织间质状态之间的关系。进一步对4个表达模块和差异基因的交集分析,功能富集分析,和PPI分析发现,NEB、DMD、IGF1、KRAS可作为BPH和正常前列腺组织基因分型的hub基因。其中IGF1经由雄激素受体通路诱导,主要通过旁分泌刺激前列腺间质细胞和前列腺上皮细胞的病理性增殖,特别是成纤维细胞的增殖[24]。间质细胞自身表达IGF1受体,因此能够被IGF1激活,经由IGF1通路,加快有丝分裂。BPH组织中KRAS基因活性抑制也同时得到了其他研究团队的验证,其在前列腺中的激活状态也收到雄激素受体通路的调控[25]。而NEB和DMD基因在BPH疾病中的功能和作用目前尚无文献报道。
综上所述,本研究对BPH和正常前列腺组织的转录组数据进行生物信息学分析发现,BPH组织中间质细胞成分显著高于正常前列腺组织。BPH组织存在较多的成纤维细胞和较少的平滑肌细胞,这可能和前列腺的发生发展相关。本研究也建立了4个基因的BPH诊断模型,这些信息对BPH的分子生物学机制研究及药物研发可能具有一定的参考价值。
