基于生物信息学分析的圆锥角膜相关Hub基因及其通路的鉴定
2021-03-02朱叶谭秋凡巩倩文胡晓建陈世豪
朱叶,谭秋凡,巩倩文,胡晓建,陈世豪
1.温州医科大学附属眼视光医院 屈光手术中心,浙江 温州 325027;2.义乌市妇幼保健院 五官科,浙江 金华 322000
圆锥角膜(keratoconus,KC)是一种多因素病因的双侧角膜变薄性疾病,以进行性角膜变薄变陡,不规则散光,视力下降,角膜前突为主要表现[1]。已有研究表明,遗传因素在KC发病过程中发挥重要作用[2]。虽然多年来,KC的众多候选基因已被确定,但这些孤立的基因尚不能解释疾病的复杂机制[3]。 目前,微阵列分析作为在基因组水平上获得基因表达数据的一种有前景的工具,已被广泛应用于疾病的分子发生和发展研究中[4]。随着生物信息学技术的发展,基于网络的方法使疾病机制的研究更加深入。SHARIF等[5]利用蛋白相互作用(protein- protein interaction,PPI)网络分析鉴定了EGFR、NEDD4、SNTA1、LGALS3BP、HSPB1、SDC2、Mme和HIF1a等多种网络节点,这些节点被称为关键(Hub)基因(蛋白质),在维持网络结构和提供生物系统的关键信息方面起着关键作用[6-8]。本研究在网络分析的基础上[9],进一步使用ClueGO使基因矩阵中最重要的条目在网络中可视化,为观察他们之间的相互关系提供了更加深入的视角,同时从分子水平上进一步了解KC的发生发展,并探索了诊断、预后和药物靶点的潜在候选的生物标志物。
1 材料和方法
1.1 微阵列分析数据 从GEO数据库下载GSE77938基因表达谱。GSE77938基于Affymetrix GPL18460 平台(Affymetrix Human,Illumina HiSeq 1500),由 KABZA等[10]提交。GSE77938数据集包含50个样本,其中有25个KC样本和25个正常对照样本(下载地址:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE77938)。
1.2 差异表达基因(differentially expressed genes,DEGs)的数据预处理与鉴定 用R语言(v.3.3.2)对原始微阵列数据进行预处理。基于平台 上的注释信息,将探针IDs转换成相应的基因名。当 多个探针对应于同一基因时,计算平均表达值来表示基因表达水平。样本分为KC组和对照组。然后,采用线性模型进行微阵列分析(Bioconductor的limma包)以识别DEGs。以|log2fold change(FC)| >0.992并且P<0.05作为DEGs的临界值。
1.3 DEGs的功能富集分析 基因本体论(GO,http://www.geneontology.org/)是一种常用的对基因和基因产物进行注释和识别特征生物的方法。京都基因和基因组百科全书(KEGG,http://www.genome.jp/kegg/Pathway.html)通路富集分析是一个包含生物化学通路的生物信息学数据库。注释、可视化和集成发现的数据库(DAVID,http://david.abcc.Ncifcrf.gov/)在线软件,是从大量基因中提取生物意义的分析工具。DAVID分别用于上调和下调DEGs的GO富集和KEGG通路富集分析。具有显著差异的功能和通路的临界值为P<0.05,计数(在特定功能或途径项中富集的基因数)>2。
1.4 PPI网络的构建及子网络分析 检索相互作用基因的搜索工具(STRINGv-10.0,http://stringdb.org/)数据库是用来评估PPI信息的在线工具。为了评价DEGs之间的相互作用关系,将DEGs映射到STRING上,并从蛋白质水平筛选DEGs之间的相互作用关系。然后,在联合评分>0.4 的条件下,分别构建了上调和下调DEGs的PPI网络。然后,利用 Cytoscape软件构建PPI网络。采用插入式分子复合物检测技术(MCODE)对Cytoscape中PPI网络的子网络进行了筛选。选择阈值节点密度截断值为1、节点得分截断值为0.2、k核为3、最大深度为100的聚类进行进一步分析。采用ClueGO插件对Hub基因列表进行可视化GO和KEGG分析。
2 结果
2.1 DEGs的鉴定 分析的样本为25个KC样本和25个正常样本。根据筛选标准,KC患者角膜共鉴定出1 547个DEGs,其中上调基因1 103个,下调基因444个(见图1)。

图1 DEGs的火山图
2.2 GO富集分析 上调和下调表达基因富集的前5个GO项见图2。GO分析结果表明,上调的DEGs在生物过程(biological process,BP,1 220)、分子功能(molecular function,MF,102)和细胞成分 (cell composition,CC,102)中均显著富集。在BP中,这些基因显著参与免疫应答(P<0.001)、免疫系统过程(P<0.001)和防御应答(P<0.001)。MF和CC的最显著项分别是抗原结合(P<0.001)和质膜(P<0.001),而下调的DEGs在BP(99)、MF(47)和CC(64)中也有富集。BP、MF和CC的最显著富集项分别为感知觉(P<0.001)、门控通道活性(P<0.001)和膜内成分(P<0.001)。
2.3 KEGG通路分析 上调和下调基因的KEGG通路见图3。上调的DEGs在72条通路中显著富集,其中前5个最具有显著差异的通路分别为细胞因子-细胞因子受体相互作用、趋化因子信号途径、移植物排斥反应、类风湿关节炎和移植物抗宿主病。而下调的DEGs在8条通路中显著富集,其中富集最显著的前5条通路为味觉传导、cAMP信号途径、谷氨酸突触、5-羟色胺能突触和2型糖尿病。

图2 KC相关DEGs的前5位GO功能富集分析
2.4 PPI网络和子网络 基于STRING数据库的信息,分别构建了上调和下调DEGs的PPI网络。在上调DEGs的PPI网络中,共有602个节点和9 903个相互作用,识别出三个聚类(见图4)。Hub基因(度>150)为TNF(235)、JUN(231)、IFNG(216)、PTPRC (197)、ICAM1(194)、FOS(193)、IL6(192)、CXCL8 (178)、LCK(165)、CSF2(163)、MMP9(162)、ITGAX (152)、CD40LG(152)和IL10(151),对于下调的DEGs,共有143个节点和319个相互作用(见图5)(度>150)。
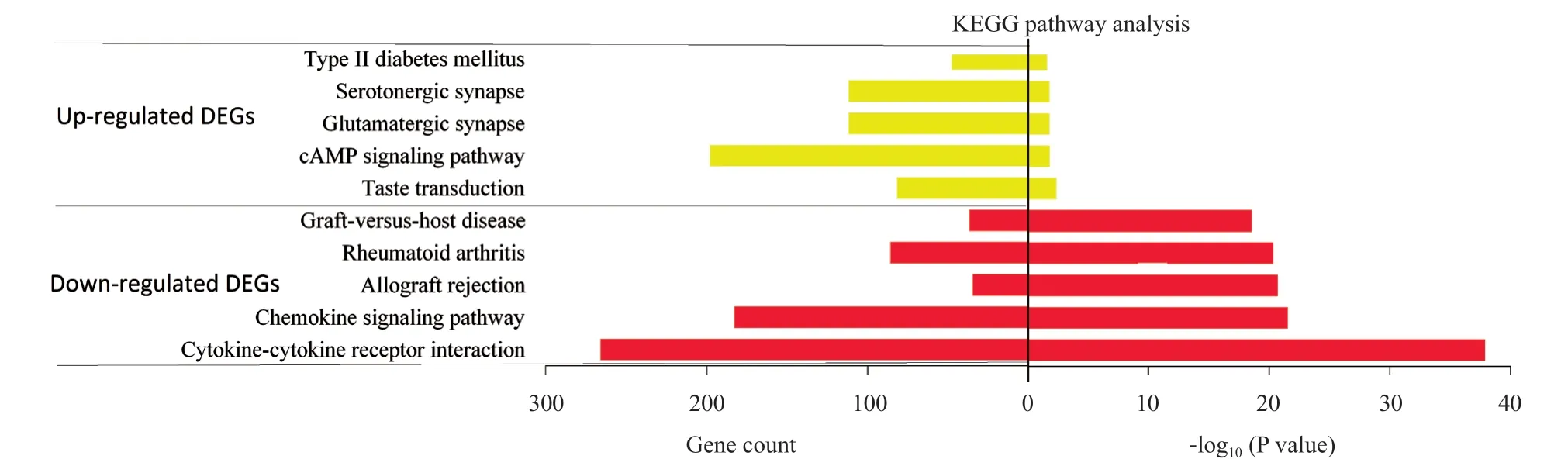
图3 KC相关DEGs的前5位KEGG通路分析

2.5 Hub基因的ClueGO分析 通过ClueGO分析,将Hub基因和通路网络进行可视化展示。结果表明,这些Hub基因显著富集在6个GO项中(见图6)。最显著的是骨髓白细胞分化的阳性调节(P <0.01)、STAT蛋白酪氨酸磷酸化的阳性调节(P<0.01)、平滑肌细胞增殖(P<0.01)、平滑肌细胞增殖的调节 (P<0.01)、细胞对脂多糖的反应(P<0.01)以及趋化因子生物合成过程的调节(P<0.01)。而Hub基因的KEGG途径在3个通路显著富集(见图7)。最显著的是NF-κB信号通路(P<0.01)、原发性免疫 缺陷(P<0.01)和T细胞受体信号通路(P<0.01)。

图5 PPI网络用于表示下调表达的DEGs
3 讨论
KC是一种异质性退行性疾病。其病因和潜在的病理机制尚不清楚,但环境和遗传因素都被认为是导致该疾病发展的相关因素[11-12]。最近,12项不同的KC研究已经确定了至少17个基因座,表明KC可能是由不同家族的许多不同基因突变引起的[13],因此了解KC的分子机制和关键基因对于诊断和治疗具有重要意义。本研究从GSE77938中提取数据,利用生物信息学分析鉴定KC和正常对照之间1 103个上调和444个下调的DEGs。为了更好地理解DEGs的相互作用进一步进行了GO功能富集分析、KEGG通路分析和PPI网络分析。通过构建PPI并应用MCODE和ClueGO插件发现了一些Hub基因和关键通路,为KC的治疗研究提供了新的思路。

构建了上调和下调DEGs的PPI网络后,发现最高度的基因(度>20)均处于上调DEGs中,充分提示上调的DEGs具有KC的生物活性。GO分析表明,上调的DEGs在BP方面主要参与免疫应答、免疫系统过程和防御应答的调控等过程。MF和CC主要表现在抗原结合、受体结合、细胞因子活性和质膜、细胞外周,这些结果表明,该病的病因与免疫反应可能存在根本的联系或相关性。而过去研究发现KC患者血清免疫球蛋白E水平较高,免疫稳态受到损害等表现也从侧面确认了变态反应和KC之间的联系 性[14-16]。此外,上调DEGs富集的KEGG通路主要包括细胞因子-细胞因子受体相互作用和趋化因子信号途径,这与角膜上皮细胞和基质细胞能够合成趋化因子、细胞因子及其受体,以及这些分子在角膜伤口愈合和炎症反应中能够发挥作用的认识是一致的[17]。WHEATER等[18]报道在其他类型的角膜疾病中,如大疱性角膜病变,这些通路也发生了变化。
通过用上调的DEGs构建PPI网络,并应用MCODE和ClueGO,发现了几个重要的通路(骨髓白细胞分化的阳性调节,STAT蛋白酪氨酸磷酸化的阳性调节,平滑肌细胞增殖,平滑肌细胞增殖的调节,细胞对脂多糖的反应、趋化因子生物合成过程的调控、NF-κB 信号通路、原发性免疫缺陷和T细胞受体信号通路)和14个Hub基因,这些通路和基因可能是诊断KC的有效途径。
本研究相比KABZA等[10]的研究最大的不同也是本研究的主要创新点在于,本研究不仅仅分析了基因的表达差异,同时结合了GO进行数据分析(KABZA等[10]的研究并未使用GO分析方法),且发现了关联基因TNF、JUN、IFNG、PTPRC、ICAM1、FOS、IL6、CXCL8、LCK、CSF2、MMP9、ITGAX、CD40LG和IL10。在这些发现的与KC相关联的基因中,TNF、IFNG、PTPRC、ICAM1、FOS、CXCL8、LCK、CSF2,ITGAX基因之前未见报道。
本研究中,肿瘤坏死因子(tumor necrosis factor,TNF)基因在网络中显示出最高的度(232)。TNF作为一种主调节因子在平衡细胞存活、凋亡和坏死中的作用已经在各种细胞类型和组织中被广泛研究[19]。有研究发现单侧KC患者和对侧有亚临床疾病的患者,双眼IL-6和TNF-α水平均升高,但TNF-α只在KC眼中显著升高[20]。CHEUNG等[21]发现,TNF-α能够触发角膜细胞凋亡,以及增加胶原蛋白的更新率。此外,POULIQUEN等[22]认为TNF-α可能调节一个涉及纤溶酶系统的蛋白酶级联反应,最终导致KC细胞外基质的变化。
据报道,PPI网络中的IL6、MMP9、IL10等基因在KC患者以及角膜下壁变薄病例(类似KC)的泪液中也有明显升高。这是一个非常有力的证据,同样支持角膜变薄和扩张与涉及炎症事件的细胞外基质降解有关(主要是MMP-9、IL-6和TNF-α水平升 高)[23-24]。
本研究在KC中获得了一些重要的基因和通路,但还存在一些局限性:基于生物信息学方法进行探究,结论尚未得到生物学实验的证实;研究样本量有限。进一步探讨KC的分子机制,并将分子遗传学诊断应用于临床,有待进一步地探索[25]。
综上所述,本研究提出了几个与KC相关的Hub基因,系统地介绍了与之相关的生物过程和信号传导通路,这些基因很少有被证实,但其中有许多被报道与KC有关。这些基因可作为KC治疗的分子靶点和诊断性生物标志物,应引起更多的研究关注。通过生物信息学分析揭示这些基因有可能与KC的病理机制相关,后续的研究需要利用细胞实验和动物实验进行深入的验证,为寻找新的靶点药物奠定关键的科学基础。


图7 上调DEGs中Hub基因的ClueGO分析
