载体介导杜氏肌营养不良症的基因治疗研究现状
2021-03-01张鹏程宋亚锋
张鹏程,宋亚锋
0 引言
杜氏肌营养不良症(Duchenne Muscular Dystrophy,DMD)是由肌营养不良蛋白(Dystrophin)缺失或减少引起的全身肌肉渐进性损伤和运动功能减退的致死性基因遗传病。致病基因DMD位于人体23号性染色体Xp21.1-p21.2上。基因组全长2.26Mb,包含79个外显子。由于致病基因尺寸巨大、累及人体肌肉组织庞大且遍布全身,为DMD患者治疗带来困难。目前,尚无有效的治疗方法。但随着人们对DMD的发病机制和病理变化过程逐渐深入认识,全世界对DMD患者已采用包括药物治疗、物理治疗、基因治疗、干细胞疗法和运动训练疗法等多种治疗手段。
正常DMD基因长度较长,患者除了从亲本获得性遗传导致发病之外,多数病例是由自发的新突变引起。因此不同患者之间Dystrophin蛋白缺失或减少的类型和程度不同,对机体的影响也就不尽相同。基因治疗具有一定的普适性,能从根本上改善肌肉功能。载体介导DMD基因治疗正是通过向组织或细胞内递送具有功能的基因来代替受影响的DMD基因,成为DMD基因治疗的主要研究热点之一。本文就近年载体介导DMD基因治疗综述如下。
1 背景
DMD在19世纪中期被鉴定为一种遗传性疾病。DMD患者多于2~5 岁时出现肌肉无力,伴随肌肉功能逐渐下降,最终在20~30岁死于心肺功能衰竭[1]。每5 000个新生男婴中就有1个患有DMD[2]。通过无创机械通气和心血管药物治疗与无任何干预的治疗相比,能延长DMD患者寿命[3]。糖皮质激素也能够改善DMD患者的肌肉力量和呼吸功能,提高患者独立行走能力[4-5]。但这些干预措施并没有从微观上改善患者的肌肉结构。
Dystrophin 蛋白在肌细胞骨架肌动蛋白和跨膜肌营养不良蛋白糖蛋白复合物(Dystrophin-associated glycoprotein complex,DGC)之间形成重要的机械连接,这些蛋白质共同保护肌纤维膜免受肌肉收缩期间所产生应力带来的损伤[6]。在缺乏 Dystrophin蛋白的情况下,无法募集DGC复合物,肌纤维膜变得极不稳定。导致肌纤维坏死,炎性细胞浸润。肌纤维再生以及最终肌纤维被结缔组织取代并伴有进行性肌硬化和收缩性丧失。肌肉随年龄增长因肌纤维再生能力下降而逐渐退化消失,最后累及心肌和膈肌[7]。
DMD另一种更温和、进展更缓慢的变体被称为贝克尔肌营养不良症(Becker muscular dystrophy,BMD)。一位61岁BMD患者缺失17~48号外显子,几乎是DMD一半的编码信息[8]。但该患者肌营养不良症状发展速度非常缓慢,寿命明显高于DMD患者,其原因为突变在DMD基因中的位置不同。随着人们对肌肉中DMD基因cDNA认识,Cox等[9]通过Full-length Dystrophin cDNA(约14Kb)转基因纠正mdx小鼠(DMD动物模型)肌营养不良症状的潜力,探索DMD基因治疗的可行性。结果显示,在转基因mdx小鼠肌肉中Dystrophin的表达消除了肌营养不良引起的形态学和免疫组织学症状,证明了DMD基因治疗的可行性。为适应病毒载体有限的包装能力,Phelps等[10]构建缺失17~48号外显子的mini-cDNA(约6Kb)转染mdx小鼠,发现短序列肌营养不良蛋白依旧可以改善小鼠肌营养不良症状。结果表明,病毒载体介导短序列肌营养不良蛋白基因可作为DMD患者的治疗方法之一。
2 短序列肌营养不良蛋白的优化设计
2.1 Dystrophin蛋白的结构 Dystrophin蛋白共具有 4 个结构域:氨基端(N-terminal domain,N)、中央棒状区(Central rod domain)、半胱氨酸丰富结构域(Cysteine-rich domain,CR)和C-末端结构域(C-terminal domain,C)。氨基端包含2个钙调蛋白同源分子结构,该结构域可以与胞浆内的肌动蛋白结合;中央棒状区由2 400多个氨基酸组成并构成24个同源三重螺旋重复区(Spectrin-like repeats)和4个铰链域(Hinge)以维持Dystrophin的灵活性;半胱氨酸丰富结构域和C-末端结构域与肌细胞膜蛋白结合(图1)。
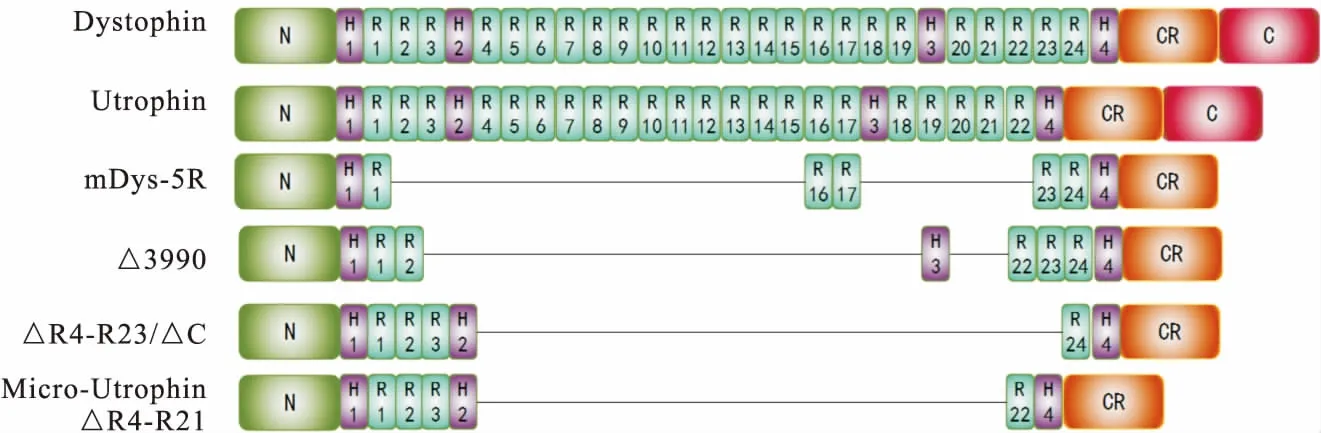
图1 Dystrophin和Utrophin蛋白结构域模式图注:N为N-terminal domain,H为Hinge,R为spectrin-like repeats,CR为cysteine-rich domain,C为C-terminal domain。△为设计短序列蛋白时去掉的部分
2.2 优化设计Micro-Dystrophin 随着生物学的不断发展,研究者成功从肌肉细胞中分离出DMD cDNA、Dystrophin蛋白,且伴随对DMD cDNA序列解读和Dystrophin结构深入认识,我们可以有目的地设计功能短序列并开展动物实验。Yuasa等[11]率先设计出3.7 kb的△DysM3基因,其包含Dystrophin蛋白的氨基端结构域,Hinge1、Hinge4,repeat1,以及半胱氨酸丰富结构域和C-末端结构域。但这种构型蛋白并没有在动物模型中产生作用。Crawford等[12]研究发现,在短序列肌营养不良蛋白设计时C-末端结构域可以删减但并不影响DGC蛋白复合物的募集形成。Harper等[13]对Dystrophin蛋白结构域进行了详细的功能分析,在保留氨基端和半胱氨酸丰富结构域的基础上,重新设计了中央棒状区的连接方式,以产生高功能的短序列肌营养不良蛋白。研究中所使用的不同短序列肌营养不良蛋白构型在小鼠体内产生的效果略有不同。结果表明,使用短序列肌营养不良蛋白的基因治疗可以预防和逆转营养不良的病理过程。
在之后的近20年内,人们不断重新设计优化spectrin-like repeats和Hinge组合以产生高功能Micro-Dystrophin蛋白[14]。
2.3 Micro-Utrophin可改善肌营养不良症状 对Dystrophin蛋白进行研究分析的同时,发现位于正常成年肌肉的神经肌肉接头处有一种与Dystrophin蛋白结构位置比较相似的蛋白,开始被称为Dystrophin-related protein(DRP),后更名为Utrophin蛋白。最终发现人体6q24基因UTRN(583.77kb)编码Utrophin蛋白(图1)。DMD患者在缺乏Dystrophin蛋白的情况下,Utrophin仍存在于肌膜中,并且Utrophin蛋白在胎儿和再生肌肉中表达。然而DMD患者体内Utrophin蛋白的表达量不足以改善患者因缺失Dystrophin蛋白所带来的影响。
关于Utrophin蛋白的高表达是否能在功能上补偿因缺失Dystrophin蛋白引起的肌营养不良症状,Tinsley等[15]为了更详细地研究Utrophin和Dystrophin之间的关系,克隆并测序了整个Utrophin cDNA,发现Utrophin和Dystrophin之间高度同源,表明它们来自一个共同的祖先基因。Rafael[16]发现在缺失dystrophin和Utrophin两种蛋白质的情况下,mdx小鼠表现出比只缺失Dystrophinb蛋白更为严重的肌肉无力并伴有关节挛缩、明显的生长迟缓和后凸畸形,这表明Dystrophin和Utrophin起互补作用。Ebihara等[17]使用含有Utrophin或Dystrophin转基因的腺病毒载体(ADV),在mdx小鼠模型中直接比较Utrophin和Dystrophin基因转移对肌营养不良功能的影响。在免疫功能较低的新生动物中,ADV-Dystrophin和ADV-Utrophin都改善了胫骨前肌的组织结构、收缩能力和肌肉抗损伤能力。然而,只有ADV-Utrophin能够明显改善免疫功能较强的成熟mdx动物肌肉收缩能力和抗损伤能力。此外,在免疫功能较强的成熟mdx小鼠中,与Dystrophin基因转移相比,Utrophin不仅具有显著的转基因持久性,还能够减少炎症发生率。Odom等[18]使用rAAV-6型病毒包装micro-Utrophin(△R4-R21,图1)在小鼠模型中具有很高的功能性,表明micro-Utrophin可以改变DMD的进程,使其更接近于非常轻微的BMD表型。
Song等[19]构建micro-Utrophin基因AAV载体,然后在新生mdx小鼠、金毛犬肌营养不良症(GRMD)幼犬和成年德国短毛犬肌营养不良症(GSHPMD)犬中进行研究。发现AAV介导的micro-Utrophin表达可以预防新生mdx小鼠的肌肉疾病,避免新生GRMD犬的免疫反应,减轻病理损伤。一项免疫原性测试中,对2只7岁的GSHPMD狗进行了并排比较。一侧胫骨肌接受micro-Utrophin载体(2.0×1012vector genome(vg)/kg),对侧肌肉注射等量的micro-Dystrophin蛋白载体。注射肌肉4周后进行活检。经H&E染色后,micro-Dystrophin蛋白处理后肌营养不良症状比较严重。与之形成鲜明对比的是,micro-Utrophin处理的肌肉中肌营养不良症状明显减弱。免疫组化染色显示micro-Utrophin蛋白表达旺盛,而micro-Dystrophin表达不旺盛。Dystrophin蛋白处理的肌肉中检测到大量的CD3+和CD8+T细胞,而Utrophin处理的肌肉中未检测到。
以上研究表明,与Dystrophin高度同源的Utrophin蛋白能够替代Dystrophin蛋白行使一定功能,缓解肌营养不良的症状。但DMD患者因为缺失DMD基因,大多不同程度缺失或无完整Dystrophin蛋白,因此会对外源Dystrophin蛋白发生免疫反应[20-21]。在这种情况下,Utrophin蛋白作为Dystrophin蛋白基因转移的替代物可能具有显著的治疗优势。
3 表达载体的构建
临床基因治疗方案最早是针对造血系统疾病。在体外经过基因转导纠正的细胞回输到患者体内以获得治疗。原则上,可以认为DMD患者的成肌细胞也可以在体外分离和处理。将纠正后的成肌细胞移植到患者肌肉中,以期恢复正常的肌肉功能。由于成肌细胞在发展为肌细胞之前可以大量增殖,单个成肌细胞或肌源性干细胞便可以纠正大量肌肉组织。但来自DMD患者自身成肌细胞的复制能力很低,极大地限制了体外细胞的培养、纠正和再转移的可能性。异源成肌细胞移植可以将正常的DMD基因带入体内,但收效甚微。且异源成肌细胞受到供体细胞免疫排斥和大规模体外培养后发展为肌细胞潜能丧失的限制。因此,利用表达载体的构建直接向组织或细胞递送目的基因,就能够克服体外基因转导复制率低和异源成肌细胞移植免疫排斥的问题。
只有少数已知的病毒载体,包括腺病毒(Adenovirus)和疱疹病毒(Herpesviruses),能够携带长达14 kb的Dystrophin cDNA和一个启动子。但这类病毒复制能力低、缺乏安全性,因此不被作为载体选用。AAV载体的携带能力为5 kb,不能包装14 kb的Dystrophin cDNA。在转基因小鼠中进行的研究表明,高度功能性的短序列肌营养不良蛋白基因可以小于4 kb[22]。直接肌肉注射AAV载体可在肌肉中产生高水平和持续的基因转移。但肌肉注射只能局部感染一小块肌肉,无法全身系统性传递。并且肌肉注射往往会引发更多的炎症,比血管输送方法更易诱发T细胞免疫反应。Gregorevic等[23]使用AAV-6型载体包装目的基因注入血管,在提高外周微血管的急性通透性下,高剂量病毒载体[1014(vg)/kg]可以转染所有成年老鼠的骨骼肌。单次静脉注射AAV-6、AAV-8和AAV-9可转染啮齿动物和大型哺乳动物全身肌肉[24-25]。这些理想的特征促进了AAV作为肌肉基因治疗载体的发展。
在不同AAV载体转染效率的研究中,AAV-8在小鼠中肌肉转导效率较高,AAV-rh74显示了肝脏偏好,AAV-9显示了心脏偏向性[26-27]。在正常和受影响的狗中所进行的几项研究中测试了高剂量的全身AAV-8和AAV-9给药。研究表明,AAV介导传递的基因存在于身体的所有肌肉中,但肝脏的含量最高[28-29]。AAV-9在小鼠中具有心脏偏向性的,在犬中传导心脏的效率要低得多[30]。重组腺相关病毒(rAAV)具有安全性高、免疫原性低、宿主范围广、能介导外源基因在动物体内长期稳定表达等特点[31],成为基因治疗领域最具应用前景的载体之一。
4 大型动物实验和临床治疗研究
由于对小鼠的研究已较为成功并显示出重要的前景,载体介导DMD基因治疗研究转向了对更大的犬类DMD模型。DMD犬类模型不仅可以评估基因治疗后肌肉伸缩性,还可以对表达载体的潜在免疫反应进行更灵敏的测试。Yue等[32]在幼年DMD犬短暂或持续的免疫抑制下,对3只2个月大的狗进行AAV-9型病毒载体携带micro-Utrophin基因(1.92~6.24×1014vg/kg)的静脉注射。DMD犬对注射耐受良好,未观察到不良反应。在骨骼肌、膈肌和心脏中观察到广泛的蛋白表达并存在肌肉组织学的改善。证明了AAV载体介导DMD基因治疗在幼年DMD大型哺乳动物中是安全有效的。
Le Guiner等[33]在共12只金毛猎犬肌营养不良症(GRMD)犬上进行研究。使用rAAV-2型和rAAV-8型载体携带Micro-Dystrophin表达基因转染GRMD局部和全身肌肉的治疗效果是有效的。局部转染肢体肌肉组织诱导高水平的Dystrophin蛋白表达,并显著改善肌肉收缩能力。全身静脉给药后未发现明显不良免疫后果,并有效减轻肌营养不良症状。以上研究表明,选择合适病毒载体(特别是可选择的病毒衣壳血清型)加上免疫系统中不活跃的短序列肌营养不良蛋白在血管传递系统下进入DMD患者体内有望实现治愈。
Mendell等[34-35]开展了载体介导短序列肌营养不良蛋白(△3990,图1)的临床合作研究,共涉及6例DMD患者。但没有患者表现出显著水平的Micro-Dystrophin蛋白表达。6例患者中,2例对肌营养不良蛋白表现出较低水平的T细胞免疫应答,另1例对AAV载体表现出明显的T细胞应答。
2017年12月发表的一篇文章为载体介导基因治疗提供了原则证明。Mendell等[36]使用单次静脉注射AAV-9载体携带目的基因[剂量2×1014(vg)/kg]治疗了患有1型脊髓性肌萎缩症(Spinal muscular atrophy type 1,SMA1)的婴儿。美国又陆续开始了3项独立的AAV介导DMD患者基因治疗试验,分别是 Solid Biosciences (NCT03368742)、Pfizer (NCT03362502)和Nationwide Children′s Hospital (NCT03375164)。此3项临床试验分别采取mDys-5R[37]、△3990[38]、△R4-R23/△C[13]3种micro-Dystrophin构型(图1)。Genethon and Sarepta Therapeutics也计划在欧洲开展临床试验。
5 未来前景思考
人体对病毒载体的免疫是一个重要问题。在没有某种形式的短暂免疫抑制的情况下,高剂量的AAV载体可诱导产生抗体,从而阻碍了使用第二剂病毒载体的能力[39-40]。如果动物研究的结果能很好地应用于人类,那么就有可能从单一剂量中获得足够水平的肌营养不良蛋白来纠正所有的肌细胞。
另一个困难是如何测量遍布全身的肌营养不良蛋白的表达和分布。目前,测量肌营养不良蛋白表达的唯一直接方法是对肌肉进行活检分析。由于活检对测试者肌肉有侵入性,并且从患者身上获得的样本量很少,这就限制了在长时间内对多个肌肉组织细胞或整块肌肉中目的基因表达效果的追踪。因此,开发可以无创监测的血清生物标志物,以及使用磁共振成像(MRI)等成像技术来跟踪肌肉结构的发展其有良好前景[41-44]。最清晰的获益指标也可以来自功能测量,其中许多已被开发用于其他DMD试验,如6分钟步行试验等[45]。
虽然动物实验显示载体介导DMD基因治疗安全有效,但能否改善人类的肌肉病变、病变改善所需时间、治疗效果的持续时间及人类能否耐受高剂量病毒载体传递等问题等均不明确。年龄和疾病进展对转导效率是否有影响?T细胞和/或B细胞的免疫反应是针对肌营养不良蛋白还是载体衣壳蛋白?数年后,当抗营养不良蛋白的表达低于治疗所需的阈值时,能否重新对载体进行调节?这都需要进一步研究。
6 结论
利用载体介导DMD基因治疗最初在mdx小鼠中开发的方法被证明是安全的,并且在小鼠的寿命中很大程度上消除了肌营养不良的病理生理学问题。在针对DMD的犬类模型中也观察到了类似的结果,而且AAV载体已被证明在针对其他遗传疾病的非人类灵长类动物研究和临床试验中是安全的。载体介导基因具有巨大的治疗潜力,因为它能够解决DMD和BMD患者不能产生正常肌营养不良蛋白的根本问题。
